
To evaluate the bacterial microbiome found in tracheostomy cannulas of a group of children diagnosed with glossoptosis secondary to Robin Sequence (RS), and its clinical implications.
MethodsPediatric patients were enrolled in the study at the time of the cannula change in the hospital. During this procedure, the removed cannula was collected and stored for amplicon sequencing of 16s rRNA. DNA extraction was performed using DNeasy PowerBiofilm Kit (QIAGEN® ‒ Cat nº 24000-50) while sequencing was performed with the S5 (Ion S5™ System, Thermo Fisher Scientific), following Brazilian Microbiome Project (BMP) protocol.
ResultsAll 12 patients included in the study were using tracheostomy uncuffed cannulas of the same brand, had tracheostomy performed for over 1-year and had used the removed cannula for approximately 3-months. Most abundant genera found were Aggregatibacter, Pseudomonas, Haemophilus, Neisseria, Staphylococcus, Fusobacterium, Moraxella, Streptococcus, Alloiococcus, and Capnocytophaga. Individual microbiome of each individual was highly variable, not correlating to any particular clinical characteristic.
ConclusionThe microbiome of tracheostomy cannulas is highly variable, even among patients with similar clinical characteristics, making it challenging to determine a standard for normality.
Tracheostomy is a surgical procedure that creates a shortcut for the air to reach the lungs.
It establishes a communication (or tunnel) between the cervical skin and the trachea, which is maintained pervious by a cannula.
Indications for tracheostomy in children nowadays are mainly related to chronic obstruction of upper airways (e.g., laryngeal stenosis or vocal fold paralysis), management of mechanical ventilation, and treatment of bronchopulmonary aspiration.1–3 Another obstructive airway pathology with potential necessity for tracheostomy is glossoptosis. Glossoptosis is defined as the posterior collapse of the base of the tongue resulting in varying degrees of respiratory obstruction. It may occur as a consequence of mandibular hypoplasia (Robin Sequence), but it also occurs in patients with neurological disorders related to hypotonia.4–6
A few days after a tracheostomy is performed, its pathway maturates, resulting in a new microbiological environment. Air goes directly to the lungs without be filtered by the upper airways, and mobilization of tracheal secretions is altered, as well as patients physiological defense systems.7–10 The cannula itself induces a constant inflammatory (foreign body) reaction, capable of accumulating organic debris and facilitating bacterial colonization and biofilm formation, sometimes resulting in exacerbation of clinical conditions.11,12
Precise determination of bacteria present at the tracheostomy tube can be challenging. Culture based methods may favor specific bacteria growth over others and does not contemplate the multitude of microbials present in that environment.7,13–15
Recently, high performance next generation sequencing equipment is becoming more accessible and, added to computational evolution and expansion of bioinformatic sector, genetic sequencing costs have been lower.16 Metagenomic is a culture independent method for phylogenetic identification of bacterial communities. Referred as shotgun sequencing, whole microbial genome sequencing is performed, resulting in taxonomic and possibly function gene identification.17–20 Another culture independent genetic sequencing method for microbiome determination is amplicon sequencing. Targeting specific known genes, shorter sequences can be used for taxonomic identification, therefore lowering costs while keeping efficacy at the study of bacterial communities.21–24
This study aims to evaluate pediatric tracheotomy bacterial microbiome using amplicon sequencing of 16S rRNA gene in a specific population of children with Robin Sequence and to describe the findings amidst recent literature.
MethodsSubjects and study designThis study received a certificate of ethic appreciation from “Plataforma Brasil” numbered 93356618.0.000.5327. The study was also approved by the Ethics and Research Committee (CEP) of Hospital de Clínicas de Porto Alegre. All patients had consent and approval forms signed by parents or legal caretakers prior to inclusion. We targeted a group of patients who performed tracheostomy due to glossoptosis associated with mandibular hypoplasia (Robin Sequence), with a history or plan for osteogenic distraction.
Data and samples collectionPrior to the tracheostomy cannula exchange, which is performed at our hospital by the Otolaryngology team, caretakers were questioned on patient current clinical condition (fever, use of antibiotics, etc.) and electronic medical chart was reviewed.
At the moment of cannula exchange, patients had their cannula collected with the use of sterile techniques. The cannula was bisected longitudinally as to include both distal (in contact to trachea) and proximal (in contact to skin) portions and placed into 2 different 15 mL conical tubes (Fig. 1). These tubes were stored in a −80 °C freezer within the first two hours of collection for future DNA extraction and sequencing.25–27

At the moment of the cannula exchange, cervical skin was also examined for the presence or absence of peristomal granuloma.
DNA extraction and amplicon sequencingAll samples were processed following the protocol of the Experimental Research Center of Hospital de Clínicas de Porto Alegre. DNA extractions were conducted using DNeasy PowerBiofilm Kit (QIAGEN® ‒ Cat nº 24000-50). For every patient the content of one 15 mL conical tube was used (half of tracheostomy tube) (Fig. 1). Once removed of the −80 °C freezer, samples were kept at room temperature for 10 min to facilitate handling. Using sterile material, tracheostomy tube was sectioned into small fragments on a Petri dish and DNA extraction was performed following DNeasy PowerBiofilm Kit protocol.
Once extracted, samples quality was tested using DNA spectrophotometer NanoDrop 1000 (Thermo Fischer Scientific®).
Approximately 50 ng of DNA for each sample were submitted to amplification of the V4 region of rRNA 16S gene by PCR using the following pair of primers: 515F (5′-GTGCCAGCMGCCGCGGTAA-3′) and 806R (5′-GGACTACHVGGGTWTCTAAT-3′). In order to process multiple samples at the same reaction, primer-fusion method was used so each sample received a unique nucleotide sequence (barcode) attached to its amplified products. Sequencing was conducted using S5 (Ion S5™ System, Thermo Fisher Scientific). The resulting raw data were compacted and exported as a single FastQ file.
BioinformaticsRaw data generated from DNA sample sequencing was processed according to the Brazilian Microbiome Project (BMP) protocol,23 using scripts in QIIME and RDP for 16S rRNA gene phylogenetic representation.
Sequences were filtered based on size and quality score with removal of chimeras. RDP_GOLD identifier was used for Operational Taxonomic Unit (OTU) picking based on 97% similarity, and phylogenetic identity of represented OTU was achieved using Green Genes database. Sequences which failed to achieve 97% similarity were defined as unclassified. Quality control was performed with FASTQC.
Data processing and STF extension files were also created using QIIME (v1.9.1). Analysis was performed using STAMP (v2.1.3).
Statistical analysisPrincipal Components Analysis (PCA) plots were used to identify clusters while separating samples for different variables such as feeding route, use of antibiotics in the last 3-months and presence of granuloma.
ResultsTwelve patients with Robin Sequence and tracheostomy were included in the study. Patient characteristics are summarized in Table 1. All tracheostomy tubes collected were of the same brand (PORTEX® Blue Line® Tracheostomy Tube) and uncuffed. No complications occurred.
Clinical characteristics of the 12 patients.
| Sample | Age (years) | Time from tracheostomy (years) | Feeding route | Syndrome | Comorbidities | Recurrent LRI | Time from last cannula change (days) | Cannula size | Peristomal granuloma | ATB (day of sampling) | ATB (last 3-months) |
|---|---|---|---|---|---|---|---|---|---|---|---|
| 001 | 8.2 | 8.1 | Oral | Facio-auricular-vertebral | Yes | No | 98 | 5 | No | No | No |
| 002 | 5.4 | 5.2 | G-tube | None | Yes | No | 98 | 4.5 | Yes | No | Yes |
| 003 | 7.7 | 7.5 | Oral/G-tube | Richieri-Costa-Pereira | Yes | No | 98 | 4.5 | No | No | No |
| 004 | 2.1 | 1.9 | NG-tube | None | Yes | No | 98 | 3.5 | No | No | Yes |
| 005 | 9.2 | 9 | Oral | Stickler | No | No | 91 | 4.5 | No | No | No |
| 006 | 2.2 | 2.1 | Oral | None | Yes | No | 91 | 3.5 | Yes | No | No |
| 007 | 4 | 1.4 | NG-tube | None | Yes | Yes | 138 | 4 | No | No | Yes |
| 008 | 7.2 | 3.9 | Oral | None | Yes | No | 80 | 5 | No | No | No |
| 009 | 11.7 | 8.3 | Oral | Picnodisostosis | No | No | 98 | 4 | No | No | Yes |
| 010 | 5.4 | 5.2 | G-tube | Apert | Yes | No | 119 | 4.5 | No | No | Yes |
| 011 | 9.1 | 9 | Oral | Treacher-Collins | Yes | No | 98 | 4 | No | No | No |
| 012 | 2.2 | 2.1 | NG-tube | None | Yes | No | 91 | 3.5 | Yes | Yes | No |
LRI, Lower Respiratory Infection; ATB, Antibiotics; G-tube, Gastrostomy; NG-tube, Nasogastric Tube.
Only one patient was using antibiotics during sampling, which had been started two days before because of increasing of tracheostomy secretion, without fever or any other symptoms. Five other patients used antibiotics previously while in use of the collected tracheostomy tube, all of them finished treatment for at least one month before cannula removal.
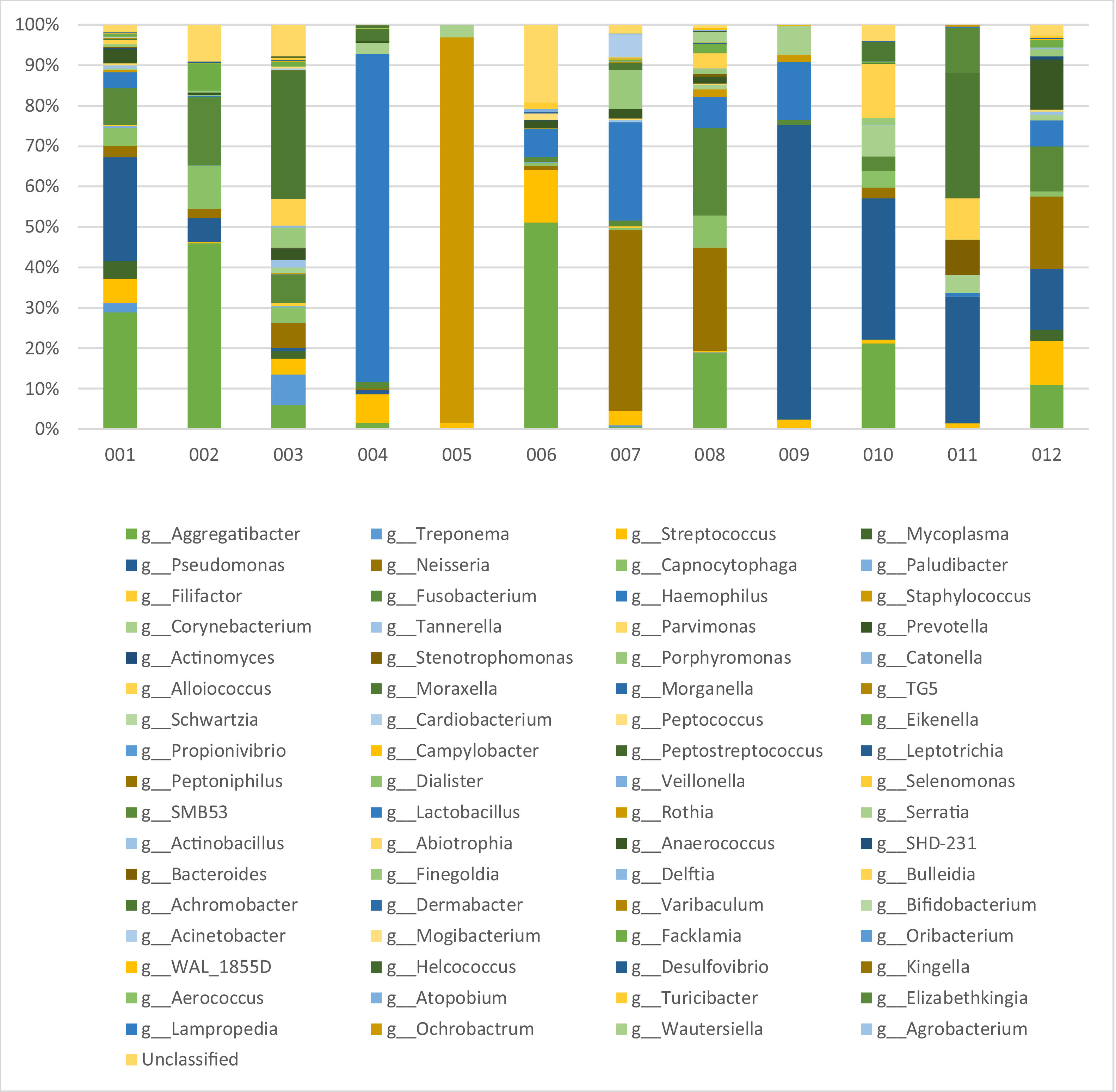
MetagenomicsDNA concentration after extraction varied between 69.65 ng/mL to 312.45 ng/mL (median = 176.84 ng/mL). After filtering and quality control, amplicon sequencing resulted in a total of 647,256 reads, ranging from 16,850 to 137,586 per sample (median = 53,938). The microbiome of each tracheostomy tube can be viewed on Fig. 2.

A total of 68 OTUs was identified at genus taxonomic level. Considering all sequences identified, most abundant OTUs included: Aggregatibacter (17%), Pseudomonas (15.9%), Haemophilus (12.4%), Neisseria (8.8%), Staphylococcus (8.3%), Fusobacterium (6.5%), Moraxella (5.8%), Streptococcus (4.6%), Alloiococcus (3%) and Capnocytophaga (2.9%). Other OTUs with lower abundance included: Corynebacterium (2.5%), Prevotella (2.5%), Porphyromonas (1.8%), Eikenella (1.2%), Treponema (0.9%), Elizabethkingia (0.9%), Stenotrophomonas (0.8%), Mycoplasma (0.8%), Achromobacter (0.5%) and Acinetobacter (0.5%) (Table 2). The 48 remaining OTUs accounted for less than 3% of all identified reads. Relative proportion of OTUs found per sample can be observed in Table 3.
Prevalence of total reads per genus on metagenomics.
| Genus | % |
|---|---|
| Aggregatibacter | 17% |
| Pseudomonas | 15.9% |
| Heamophilus | 12.4% |
| Neisseria | 8.8% |
| Staphyloccus | 8.3% |
| Fusobacterium | 6.5% |
| Moraxella | 5.8% |
| Streptococcus | 4.6% |
| Alloiococcus | 3.0% |
| Capnocytophaga | 2.9% |
| Corynebacterium | 2.5% |
| Prevotella | 2.5% |
| Porphyromonas | 1.8% |
| Eikenella | 1.2% |
| Treponema | 0.9% |
| Elizabethkingia | 0.9% |
| Stenotrophomonas | 0.8% |
| Mycoplasma | 0.8% |
| Achromobacter | 0.5% |
| Acinetobacter | 0.5% |
| Others | 2.4% |
| Total | 100% |
Genera proportion per sample.
| 001 | 002 | 003 | 004 | ||||
|---|---|---|---|---|---|---|---|
| Genus | % | Genus | % | Genus | % | Genus | % |
| Aggregatibacter | 29.48 | Aggregatibacter | 50.67 | Moraxella | 34.42 | Haemophilus | 81.27 |
| Pseudomonas | 26.33 | Fusobacterium | 18.69 | Treponema | 8.30 | Streptococcus | 6.98 |
| Fusobacterium | 9.21 | Capnocytophaga | 11.62 | Fusobacterium | 7.49 | Moraxella | 2.95 |
| Streptococcus | 6.07 | Eikenella | 7.33 | Alloiococcus | 7.15 | Corynebacterium | 2.66 |
| Capnocytophaga | 4.36 | Pseudomonas | 6.69 | Neisseria | 6.75 | Aggregatibacter | 1.58 |
| Mycoplasma | 4.32 | Neisseria | 2.44 | Aggregatibacter | 6.42 | Fusobacterium | 1.54 |
| Haemophilus | 4.05 | Prevotella | 0.72 | Porphyromonas | 5.55 | Pseudomonas | 1.28 |
| Neisseria | 2.87 | Porphyromonas | 0.59 | Streptococcus | 4.11 | Achromobacter | 0.57 |
| Prevotella | 2.81 | Haemophilus | 0.40 | Capnocytophaga | 4.08 | Prevotella | 0.43 |
| Treponema | 2.43 | Leptotrichia | 0.31 | Mycoplasma | 2.19 | Neisseria | 0.27 |
| Others | 8.07 | Others | 0.54 | Others | 13.54 | Others | 0.47 |
| 005 | 006 | 007 | 008 | ||||
|---|---|---|---|---|---|---|---|
| Genus | % | Genus | % | Genus | % | Genus | % |
| Staphylococcus | 95.23 | Aggregatibacter | 63.12 | Neisseria | 45.55 | Neisseria | 25.77 |
| Corynebacterium | 3.09 | Streptococcus | 16.15 | Haemophilus | 24.89 | Fusobacterium | 21.78 |
| Streptococcus | 1.54 | Haemophilus | 8.68 | Porphyromonas | 9.82 | Aggregatibacter | 18.99 |
| Anaerococcus | 0.05 | Peptococcus | 1.87 | Acinetobacter | 5.89 | Capnocytophaga | 7.95 |
| Finegoldia | 0.03 | Selenomonas | 1.78 | Streptococcus | 3.61 | Haemophilus | 7.89 |
| Alloiococcus | 0.01 | Prevotella | 1.66 | Prevotella | 1.90 | Alloiococcus | 3.77 |
| Facklamia | 0.01 | Fusobacterium | 1.55 | Moraxella | 1.78 | Serratia | 2.76 |
| Actiomyces | 0.01 | Capnocytophaga | 1.14 | Fusobacterium | 1.42 | Eikenella | 2.34 |
| Abiotrophia | 0.01 | Neisseria | 1.10 | Eikenella | 0.59 | Staphylococcus | 1.72 |
| Peptoniphilus | 0.01 | Veillonella | 1.08 | Aggregatibacter | 0.55 | Porphyromonas | 1.28 |
| Others | 0.01 | Others | 1.87 | Others | 4 | Others | 5.75 |
| 009 | 010 | 011 | 012 | ||||
|---|---|---|---|---|---|---|---|
| Genus | % | Genus | % | Genus | % | Genus | % |
| Pseudomonas | 72.83 | Pseudomonas | 36.42 | Pseudomonas | 31.18 | Neisseria | 18.44 |
| Haemophilus | 14.25 | Aggregatibacter | 22.11 | Moraxella | 30.10 | Pseudomonas | 15.33 |
| Corynebacterium | 7.25 | Alloiococcus | 13.71 | Elizabethkingia | 11.08 | Prevotella | 12.01 |
| Streptococcus | 2.47 | Corynebacterium | 8.20 | Alloiococcus | 10.14 | Fusobacterium | 11.36 |
| Staphylococcus | 1.64 | Achromobacter | 5.05 | Stenotrophomonas | 8.64 | Aggregatibacter | 11.29 |
| Fusobacterium | 1.19 | Capnocytophaga | 4.23 | Corynebacterium | 4.51 | Streptococcus | 11.04 |
| Anaerococcus | 0.10 | Fusobacterium | 3.60 | Streptococcus | 1.38 | Haemophilus | 6.55 |
| Porphyromonas | 0.09 | Neisseria | 2.92 | Achromobacter | 1.06 | Mycoplasma | 2.99 |
| Finegoldia | 0.05 | Porphyromonas | 1.81 | Haemophilus | 0.92 | Porphyromonas | 2.07 |
| Peptoniphilus | 0.05 | Streptococcus | 0.85 | Lampropedia | 0.36 | Eikenella | 1.89 |
| Others | 0.02 | Others | 1.1 | Others | 0.63 | Others | 7.03 |
Principal Component Analysis (PCA) plots were performed searching for clusters accounting for variables such as food intake, use of antibiotics or presence of granuloma; however, no cluster patterns were found (see Supplemental Material).
DiscussionAfter analysis of tracheostomy cannula microbiome of 12 children with glossoptosis secondary to mandibular hypoplasia and other common features (i.e., use of uncuffed tube, use of the same tracheostomy tube brand, similar length of time in use of the analyzed cannula, and tracheostomy surgical procedure performed for over a year) we can state that there is a very high microbiome variability among patients, making the establishment of procedure routines very challenging.
Aggregatibacter was the OTU with the highest number of reads in metagenomics (17% of all samples reads), present in 10 of the 12 analyzed tracheostomies tubes, representing over 10% of microbiome genera proportion in 6 samples. Belonging to the Pasteurellaceae family (also common to the Haemophilus genus) its most noticeable representative is Aggregatibacter actiomycetemcomitans, a gram-negative facultative anaerobe bacterium frequently identified in oral cavity and associated with periodontal disease. Aggregatibacter has great biofilm formation capabilities, which can have mutualistic characteristics with Fusobacterium nucleatum and Veillonella spp.28–31 Unlike our results Aggregatibacter was not found among most observed OTUs on other tracheostomy microbiome studies.32–34 Dickson et al. (2015) and Charlson et al. (2011) described that, although different environments in the same patient such as oral cavity, lower and upper airway show different bacterial communities, they are not isolated from each other. Closer environments sustain microbiomes with higher resemblance than distance environments, suggesting that these regions are not completely separate, but linked by transition areas.35,36 It is possible the abundance of Aggregatibacter and other oral cavity typical OTUs (as Neisseria, Fusobacterium, Streptococcus, Prevotella and Porphyromonas) can be correlated with patients’ disease-associated high probability of dysphagia and aspiration.5 In fact, among the 12 patients, 6 were in use of G-tube or nasogastric tube, and the others with oral feeding probably showed a high risk of silent aspiration, according to the studies of Gasparin et al. (2017) and Monasterio et al. (2004).37,38
Bacteria such as P. aeruginosa, Staphylococcus aureus, F. nucleatum and A. actinomycetemcomitans are known for development of biofilm and can create polymicrobial complexes among themselves or associated with other bacteria as Haemophilus influenzae and Veillonella spp.39–41 We found high abundances of these genus among our patients, which is believed to be expected as tracheostomy tube serves as an anchor point for these structures.
Regarding distribution, patients seemed to present with either a balanced proportion of different bacteria or a predominance of one or two genus. In four samples two genera corresponded to more than 75% of all reads (i.e., sample 005 with Staphylococcus representing 95.23% of its reads). Although these patterns could be highlighted, either group had no distinguished characteristics that could explain this finding.
Only one patient (sample 012) was receiving antibiotics during sampling, indicated for suspected lower respiratory exacerbation. The patient had increased tracheostomy secretion without fever or other symptoms. Amplicon sequencing of this sample evidenced no particular pathological bacterial genus, suggesting it was a viral or non-infectious condition. Perez-Losada shown that a microbiota altered by an acute infection tends to balance itself after four weeks. All the five patients who had received previous antibiotics had their treatment concluded more than a month before sampling. It is not clear if the use of antibiotics played a role in their microbiome at the time of the study, as their microbiome showed no particular characteristics.34
Wang et al. (2018) studied the microbiome of 4 children with tracheostomy, of whom 2 had suprastomal granuloma tissue identified by airway endoscopy.32 Both patients with granuloma tissue were in use of antibiotics and grew H. influenzae in respiratory bronchoalveolar lavage culture. Microbiome evaluation of these patients’ tracheostomy tubes through shotgun metagenomics revealed that besides H. influenzae, F. nucleatum, M. catahrralis and Streptococcus pneumoniae had higher abundances, potentially being associated with the presence of granuloma. Although airway endoscopy was not performed during our sampling, periostomal granulation tissue was observed on three patients: 002, 006 and 012. Amplicon sequencing observed 19 OTUs common to all three samples, highlighting only the ones with higher abundancy we observe: Aggregatibacter (50.7% vs. 63.1% vs. 11.3%), Fusobacterium (18.7% vs. 1.5% vs. 11.4%), Neisseria (2.4% vs. 1.1% vs. 18.4%), Prevotella (0.7% vs. 1.6% vs. 12.0%) and Haemophilus (0.4% vs. 8.9% vs. 6.5%). Granulation tissue is potentially influenced by conditions of the tracheostomy tube (oxygenation, secretion, surface), but our results show that its link to specific microbes is still not clear, as these groups of bacteria may be associated either to cause or effect. Other culture-based studies in stents have described relation of granulation tissue with Staphylococcus aureus and P. aeruginosa.42,43 According to our results, Staphylococcus and Pseudomonas populations on the three patients with identified granulomas was not characteristic, with Staphylococcus varying from none to 0.16% and Pseudomonas from 0.07% to 15.3%. It is possible that studies tend to find these bacteria easier for their capabilities on growing in culture methods and biofilm formation.
Feigelman et al. (2017) describes higher prevalence of Prevotella, Streptococcus, Veillonella, Haemophilus and Neisseria in bronchoalveolar lavages of healthy individuals without tracheostomy.44 Our samples showed a mixture of these genera intertwined with potentially pathological bacteria. As patient’s microbiome had variable presentations, they did not correlate to clinical characteristics studied (feeding route, use of antibiotics or presence of granuloma). Recent literature suggests that infections are more related to a shift in community balance than the introduction of a new bacteria, and a static microbiome analysis may not be sufficient to understand correlations between network patterns and its causes or consequences.43,44
More studies are needed in order to understand bacterial community networks and how they interact among themselves, with the environment and what the clinical repercussions are.
ConclusionOur study describes the use of 16s RNA metagenomics for bacterial microbiome identification present on tracheostomy cannulas of a specific group of children.
Although some bacterial genera demonstrated greater abundance (e.g., Aggregatibacter, Pseudomonas, Haemophilus, Neissera, Staphylococcus, Fusobacterium and Moraxella), composition of patients’ individual microbiome showed high variability, even with similar clinical characteristics.
Despite the descriptive nature of this study, we hope to create foundations for more complex questions regarding microbiology interactions of these patients.
FinancingThis Project was funded by Fundo de Incentivo à Pesquisa e Eventos (FIPE) from Hospital de Clínicas de Porto Alegre.
Conflicts of interestThe authors declare no conflicts of interest.
The following is Supplementary data to this article:
Peer Review under the responsibility of Associação Brasileira de Otorrinolaringologia e Cirurgia Cérvico-Facial.


