
Paragangliomas são tumores benignos, de crescimento lento, que surgem a partir de células da crista neural embrionária. Esses tumores neuroendócrinos têm origem principalmente a partir das glândulas suprarrenais em 90% dos casos, sob a forma de feocromocitoma. Apenas 5 a 10% são extrassuprarrenais e podem estar localizados em qualquer lugar entre o pescoço e a região pélvica, ao longo da distribuição do sistema nervoso simpático.1
Apenas 3% dos paragangliomas extra‐adrenais são encontradas na região da cabeça e pescoço. O local mais prevalente é corpo carotídeo, que dá origem ao quimiodectoma clássico. Outros locais são a região tímpano‐jugular, a região vagal, a traqueia, a língua, a laringe, a hipófise, a glândula pineal e a região orbital.2 Uma recente revisão identificou apenas 25 casos como esses em todo o mundo e 12 deles estavam na cavidade nasal.3 Nesse local, a literatura mostrou apenas três casos que tinham origem no septo nasal.4 Foi discutida a apresentação e o protocolo de conduta em um paciente de 15 anos diagnosticado com paraganglioma do septo nasal, com revisão da literatura.
Relato de casoUm jovem de 15 anos procurou nosso centro de atendimento com história de obstrução nasal esquerda gradualmente progressiva durante dois meses, com história de sangramento recorrente na narina esquerda. Não havia história de sangramento em quaisquer outros locais ou de aparecimento constante de hematomas. À rinoscopia anterior uma massa única, polpuda e de superfície lisa foi visualizada, preenchia a cavidade nasal esquerda, atingia a extremidade anterior da concha inferior e desviava o septo nasal para o lado direito. A massa era firme, dolorosa, sensível e sangrante ao toque. A rinoscopia posterior não revelou qualquer extensão coanal. À endoscopia nasal rígida, a massa foi visualizada fixada à região posterossuperior do lado esquerdo do septo nasal.
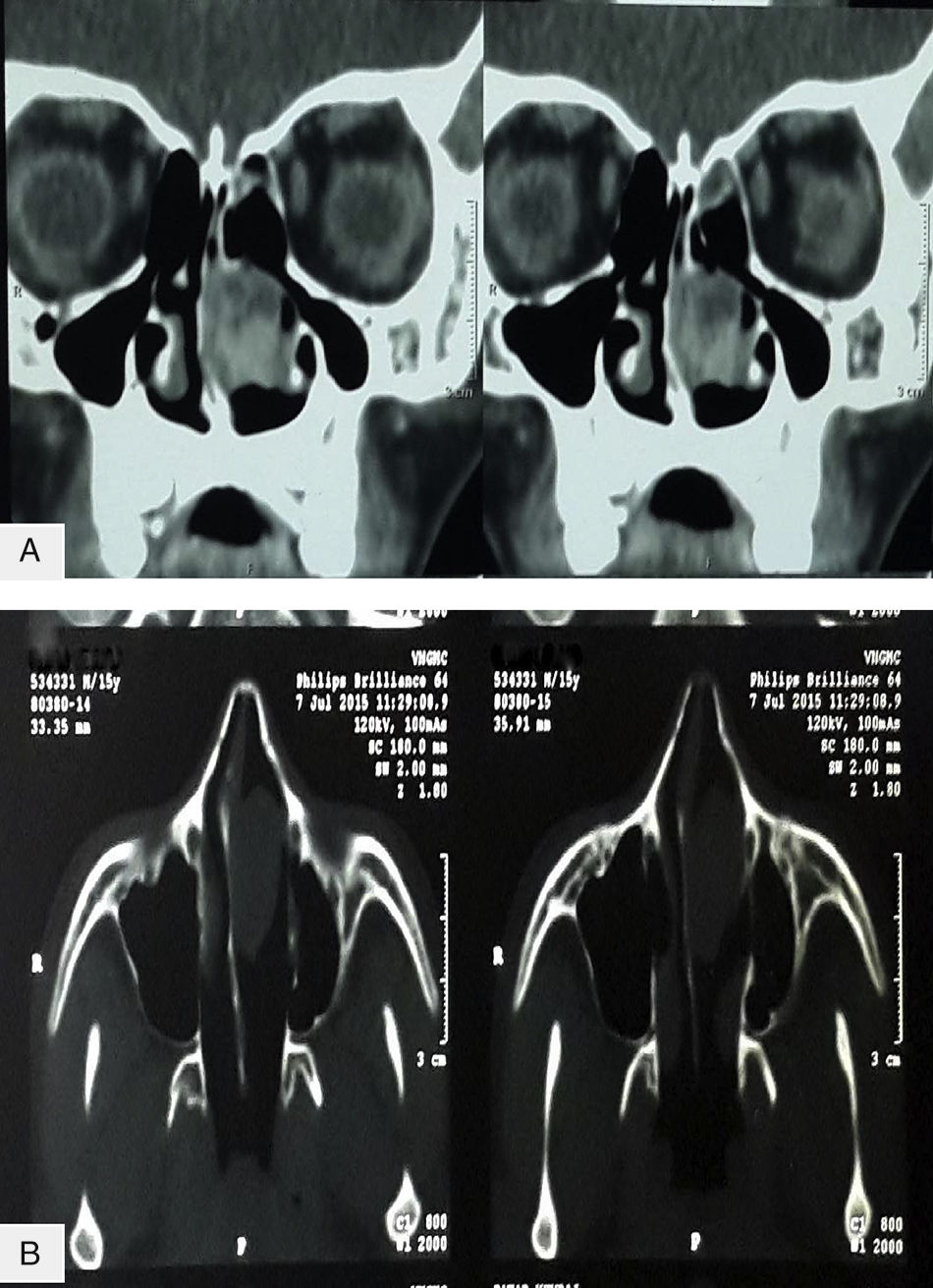
Hemograma completo, parâmetros bioquímicos e perfil de coagulação estavam dentro dos limites normais. A tomografia computadorizada (TC) dos seios paranasais mostrou uma lesão homogênea de tecidos moles que media 41×32×24mm na cavidade nasal esquerda. Todos os seios apresentavam‐se desobstruídos. Não foi identificada erosão óssea na tomografia computadorizada. O estudo com contraste mostrou realce heterogêneo, indicava um tumor vascular (fig. 1).
 e (B) axial, mostra uma lesão com densidade de tecido mole na fossa nasal esquerda, com realce heterogêneo pós‐contraste.")
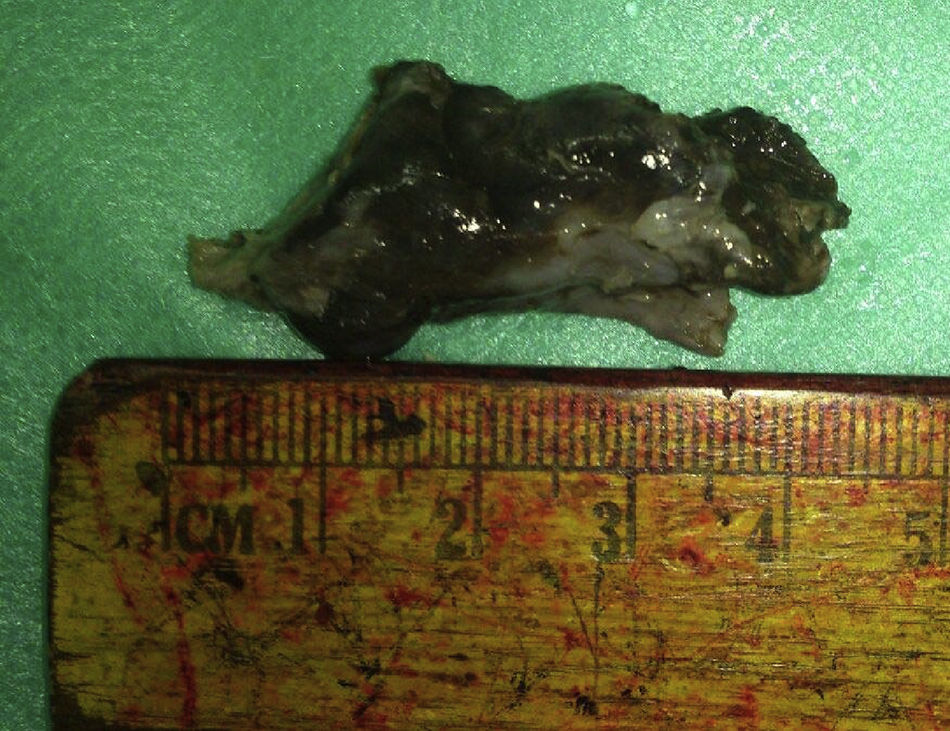
Considerando a vascularização do tumor na tomografia computadorizada, a idade e o sexo do paciente, suspeitou‐se de angiofibroma nasal e, portanto, a biópsia não foi feita. O paciente foi encaminhado para a excisão endoscópica do tumor nasal sob anestesia geral, após obtenção do consentimento livre e informado. A lesão foi removida completamente por via endoscópica regular, com a retirada de uma porção de pericôndrio septal (fig. 2). O procedimento operatório e o período de recuperação transcorreram sem intercorrências.

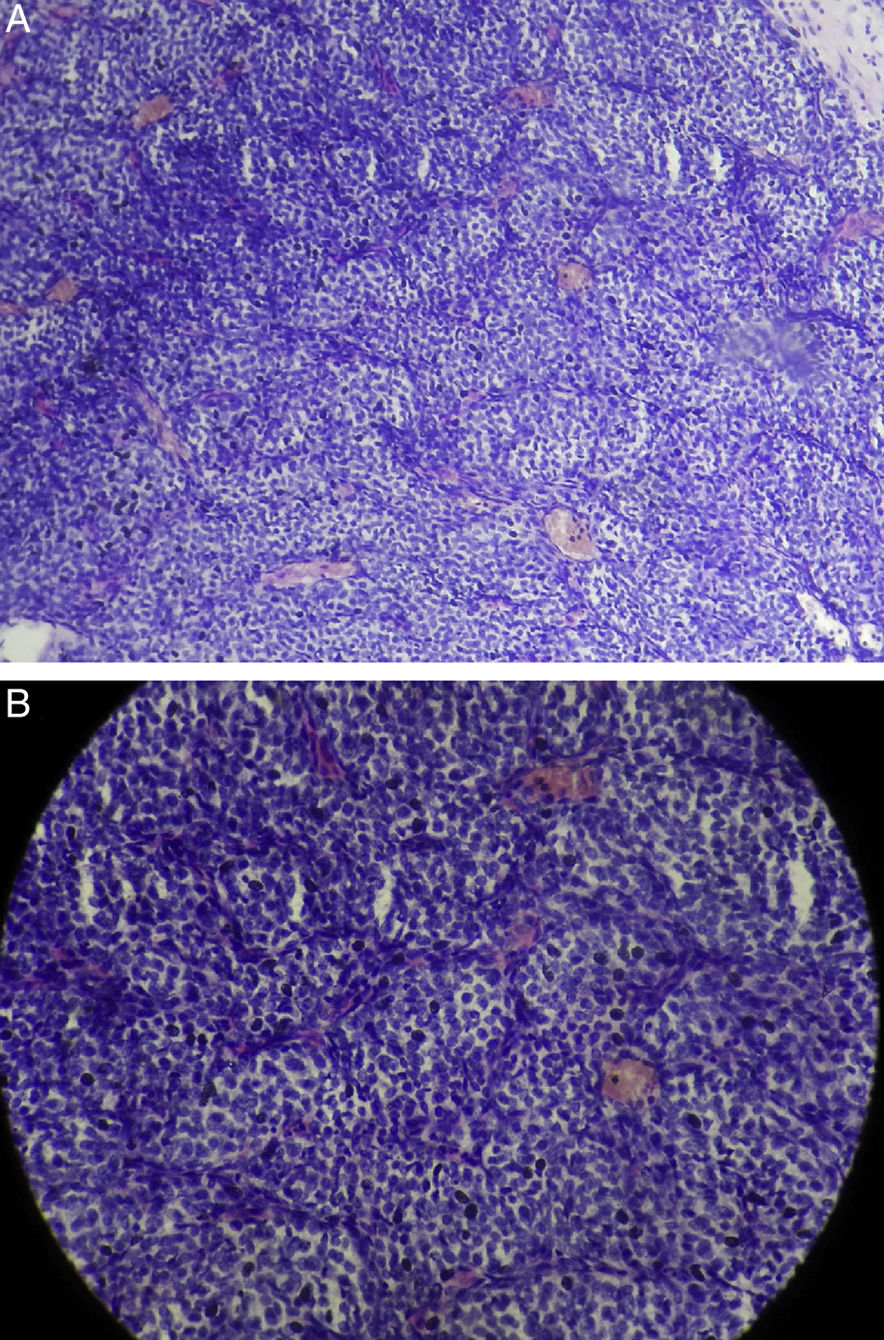
O exame histopatológico do espécime excisado mostrou células tumorais redondas e ovais, com citoplasma pálido moderado com núcleos vesiculares redondos e cromatina dispersa. As células tumorais estavam dispostas em ninho ou padrão zellballen clássico, separadas por estroma de tecido conjuntivo vascularizado. Não havia figuras de mitose ou atipia celular (fig. 3). As células tumorais mostraram imunorreatividade forte e difusa para sinaptofisina e focalmente para a proteína S100 e negativa para cromogranina, CD 10 e EMA. O diagnóstico de paraganglioma foi confirmado. Um acompanhamento regular, por um ano, não mostrou sinais de recorrência.
Discussão, Baixa magnificação; (B), Alta magnificação.")
Feocromocitoma é geralmente um tumor funcional e os pacientes apresentam sintomas de excesso de catecolaminas; mas paragangliomas da região da cabeça e pescoço são tumores não funcionais ou não cromafins.5 A identificação de paragangliomas no trato sinonasal é principalmente baseada no conhecimento de sua ocorrência nessa região. O conhecimento da distribuição de tecidos paraganglionares normais é de suma importância na previsão dos seus locais de origem. Também é essencial compreender que eles podem ocorrer fora do domínio do sistema nervoso simpático e parassimpático, o que explica a sua existência em locais incomuns.
A origem dos paragangliomas nasais é uma entidade controversa. Talbot et al. descreveram células paraganglionares na artéria maxilar distal em crianças natimortas, sustentaram a teoria da persistência de tecidos paraganglionares ao longo das artérias cranianas, o que poderia levar ao desenvolvimento de paragangliomas da cabeça e pescoço.6 Outros autores sugeriram que o tumor poderia ter sua origem no gânglio pterigoideo.3
Os seios nasais e paranasais são os locais menos comuns para essa neoplasia na região da cabeça e pescoço. A cavidade nasal é mais comumente afetada pelo tumor do que os seios. A parede nasal lateral, a concha média e seio etmoidal são as áreas mais atingidas.7 O septo nasal é um local muito raro para esse tumor, com poucos casos relatados na literatura.4
Paragangliomas nasais são tumores de crescimento lento, com um intervalo habitual entre o início dos sintomas e o diagnóstico final de dois anos ou mais. Os tumores tendem a ser bilaterais e multicêntricos e sua incidência é de 3 a 26% entre os pacientes com história familiar positiva, o que corrobora sua predisposição genética.8
Clinicamente, os pacientes apresentam epistaxe recorrente, leve a profusa, obstrução nasal, rinorreia e edema facial. Os tumores foram relatados em pacientes entre 8 e 89 anos, as mulheres de meia‐idade são as mais comumente afetadas. Rinoscopia anterior revela uma massa polipoide avermelhada na cavidade nasal, estende‐se até os seios; avaliação por tomografia computadorizada revela uma lesão expansível com densidade de tecido mole, visualizada na cavidade nasal e nos seios paranasais, com realce heterogêneo de contraste. Erosão óssea é vista em casos de paragangliomas malignos.9
Macroscopicamente, paragangliomas são tumores firmes e endurecidos, encapsulados, acinzentados ou rosados. Microscopicamente, o tumor é constituído por células epitelioides, também chamadas de células principais, com núcleos arredondados e citoplasma eosinofílico que forma um ninho, conhecido como padrão zellballen, cercado por uma rede de capilares rica em reticulina. Microscopia eletrônica revela a presença de grânulos neurosecretórios citoplasmáticos. As células neoplásicas mostram afinidade com o ácido crômico.1 A suspeita de malignidade é baseada em metástases a distância, já que, histologicamente, não há características distintas para diferenciar lesões malignas de benignas.3
Na análise por imuno‐histoquímica, os paragangliomas expressam numerosos marcadores e a cromogranina é ubíqua na maioria dos paragangliomas, uma vez que está presente nos grânulos secretores. Outros marcadores são sinaptofisina, enolase neurônio‐específica e proteína S‐100, essa última mostra coloração nas células de sustentação periféricas.9 Em nosso caso, a cromogranina foi negativa, já que paragangliomas extra‐adrenais geralmente não são funcionais.
Paragangliomas de cabeça e pescoço podem ser malignos em 4 a 19% dos casos e as metástases ocorrem em 9% deles. Linfonodos, pulmões e ossos são os locais com risco de metástases.10 Excisão cirúrgica completa com margem livre da doença é o tratamento de escolha. Arnes et al. mostraram uma taxa de recorrência de 10%. Alguns autores preferem a radioterapia à cirurgia, mas ela deve ser reservada como terapia adjuvante para tumores não totalmente excisados e pacientes mais velhos, ou aqueles com contraindicação para cirurgias. A quimioterapia não é eficaz para essa neoplasia e a embolização é usada ocasionalmente, apenas para reduzir a perda de sangue intraoperatória.10
A abordagem endoscópica endonasal é muito superior às abordagens tradicionais, como a rinotomia lateral e a técnica de degloving. É estética e funcionalmente superior, está associada a menor morbidade. Ela também fornece uma visão ampliada que ajuda na melhor delimitação da área onde o tumor está fixado e também na preservação das estruturas adjacentes importantes. Ela reduz o tempo de internação do paciente e não deixa cicatrizes cirúrgicas. No entanto, independentemente da abordagem, o seguimento em longo prazo é essencial, deve‐se ter sempre em mente a natureza agressiva do tumor. No nosso caso, o paciente foi seguido por um ano, sem qualquer evidência endoscópica ou radiológica de recorrência.
ConclusãoApresentamos um caso de paraganglioma do septo nasal em um paciente de 15 anos. Paraganglioma da cavidade nasal é uma entidade extremamente rara e houve apenas três relatos anteriores dessa doença no septo nasal. Concluímos que o paraganglioma deve ser incluído no diagnóstico diferencial de lesões vasculares nasais unilaterais e o septo nasal deve ser considerado como uma potencial localização, ainda que raro, para esse tumor. A excisão cirúrgica completa não está associada à recorrência. A análise por histopatologia e imuno‐histoquímica deve ser corroborada em casos duvidosos.
Conflitos de interesseOs autores declaram não haver conflitos de interesse.
Como citar este artigo: Gawarle S, Keche P, Ganguly S. Nasal septum: an extremely unusual location for head and neck paraganglioma. Braz J Otorhinolaryngol. 2019;85:667–9.
A revisão por pares é da responsabilidade da Associação Brasileira de Otorrinolaringologia e Cirurgia Cérvico‐Facial.
gology tem o prazer em homenagear os revisores


