
Mutations in the otoferlin gene are responsible for auditory neuropathy.
ObjectiveTo investigate the prevalence of mutations in the mutations in the otoferlin gene in patients with and without auditory neuropathy.
MethodsThis original cross-sectional case study evaluated 16 index cases with auditory neuropathy, 13 patients with sensorineural hearing loss, and 20 normal-hearing subjects. DNA was extracted from peripheral blood leukocytes, and the mutations in the otoferlin gene sites were amplified by polymerase chain reaction/restriction fragment length polymorphism.
ResultsThe 16 index cases included nine (56%) females and seven (44%) males. The 13 deaf patients comprised seven (54%) males and six (46%) females. Among the 20 normal-hearing subjects, 13 (65%) were males and seven were (35%) females. Thirteen (81%) index cases had wild-type genotype (AA) and three (19%) had the heterozygous AG genotype for IVS8-2A-G (intron 8) mutation. The 5473C-G (exon 44) mutation was found in a heterozygous state (CG) in seven (44%) index cases and nine (56%) had the wild-type allele (CC). Of these mutants, two (25%) were compound heterozygotes for the mutations found in intron 8 and exon 44. All patients with sensorineural hearing loss and normal-hearing individuals did not have mutations (100%).
ConclusionThere are differences at the molecular level in patients with and without auditory neuropathy.
Mutações no gene da otoferlina (OTOF) são responsáveis pela neuropatia auditiva.
ObjetivoInvestigar a prevalência de mutações no gene OTOF em pacientes com e sem neuropatia auditiva.
MétodoEstudo de casos em corte transversal sendo avaliados 16 casos índice com neuropatia auditiva, 13 pacientes com deficiência auditiva sensorioneural (DASN) e 20 indivíduos ouvintes. DNA foi extraído de leucócitos do sangue periférico e regiões do gene OTOF foram analisadas pela técnica PCR-RFLP.
ResultadosDos 16 casos índice, 9 (56%) são do gênero feminino e 7 (44%) do masculino. Dos 13 pacientes com DASN, 7 (54%) são masculinos e 6 (46%) femininos. Dos 20 ouvintes, 13 (65%) são masculinos e 7 (35%) femininos. Treze (81%) casos índice apresentam o genótipo selvagem (AA) e 3 (19%) o genótipo heterozigoto AG para a mutação IVS8-2A-G (intron 8). A mutação 5473C-G (exon 44) foi encontrada em heterozigose (CG) em 7 (44%) dos casos índice e 9 (56%) apresentam o genótipo selvagem (CC). Destes mutantes, dois (25%) são heterozigotos compostos para as mutações encontradas no intron 8 e exon 44. Os pacientes com DASN e os ouvintes não apresentam mutações (100%).
ConclusãoExistem diferenças, ao nível molecular, em pacientes com e sem neuropatia auditiva.
After the mutations in GJB2 and GJB6 gene, which account for approximately 80% of autosomal-recessive nonsyndromic hearing impairment (ARNSHI), mutations in the otoferlin gene (OTOF) are the most frequent genetic cause of deafness in children. This type of hearing loss (HL) is more complicated, considering that at an early stage it may present as auditory neuropathy (AN) that is not detected by neonatal screening based on otoacoustic emission testing.1
Auditory neuropathy is a unique type of HL in which tympanograms are normal, the acoustic reflex and the brainstem auditory evoked potentials (BAEP) are absent or severely altered, and the outer hair cell function is normal, as shown by the presence of otoacoustic emissions (OAEs). Patients with this disorder can show varied degrees of HL, in addition to severe speech comprehension impairment, which is disproportional to the degree of hearing loss.1
In 2003, the term “autosomal-recessive nonsyndromic auditory neuropathy” was defined, due not only to the audiological characteristics, but also to genetic discoveries related to mutations in the OTOF gene, which were associated with this type of hearing loss.2 The OTOF gene, located on chromosome 2 (2p23-p22), contains 48 exons and encodes the protein otoferlin, which is expressed in cochlear inner hair cells and type I vestibular hair cells.3 Since then, molecular studies have been published that allowed the identification of mutations in the OTOF gene and associated them to the autosomal-recessive nonsyndromic AN.2–12
Therefore, this study aimed to investigate the prevalence of mutations in the OTOF gene: 2416T-A (Y730X) in exon 18,3 IVS8-2-A-G in intron 8/exon 9,6 2485C-T (Q829X) in exon 22,5 5473C-G (Pro1825Ala) in exon 44,5 and 3032T-C (Leu1011Pro) in exon 26,9 in patients with AN and in patients with non-syndromic HL.
MethodsThis cross-sectional study was carried out from April 1, 2010 until July 30, 2011; of 1230 cases of nonsyndromic sensorineural hearing loss (NSHL) treated at the Hearing Loss Outpatient Clinic of the institution, 16 patients were selected (NPT group–neuropathy), of both genders, with AN. These were deemed index cases, according to the following inclusion criteria: normal otoscopy, diagnosis of NSHL by pure tone audiometry, presence of otoacoustic emissions and/or cochlear microphonic in BAEP; absence of BAEP waves or severe alteration in their morphology; normal MRI; absence of risk factors (maternal–fetal infections, meningitis, ototoxic drugs) or peripheral neuropathies. Exclusion criteria included a diagnosis of conductive or mixed hearing loss, the presence of risk factors or peripheral neuropathies, and an age >65 years.
A total of 13 patients of both genders (NSHL group) were also selected, who had received a diagnosis of NSHL and whose audiological tests did not match the clinical picture of AN and had no mutations in the connexin 26 and GJB6 genes.
For the Control group (CG group), 20 individuals of both genders were selected, with no hearing complaints, with normal otoscopy and who were unrelated to the patients in the NPT and NSHL groups.
The index cases, patients, and controls were of the same ethnicity, age ≤65 years, with no consanguinity, and from the same geographical area.
The study was approved by the Ethics Committee of the institution (Edict No. 44/2010). Genomic DNA was extracted from peripheral blood samples using the GE Illustra extraction kit – Blood Genomicprep Mini Spin Kit™ (GE Healthcare UK Ltd) according to the manufacturer's protocol.
All five mutations analyzed in the OTOF gene are identified by their positions in the NCBI Gene Bank RefSeqGene: NG_009937.1. Polymerase chain reaction/restriction fragment length polymorphism (PCR/RFLP) tests were used for the molecular analysis.
Each of the five PCR reactions was processed in a temperature cycler (Bioer Technology®, Model TC-XPG) using 25uL final reaction volume, containing the following: 1 – DNA (200–300ng); 2 – 10pmol of each primer (forward and reverse); 3 – Reagent Kit FideliTaq™ PCR MasterMix (2×) (GE HEALTHCARE®) using protocol according to the manufacturer's instructions.
The following standard cycle for the PCR reactions was used, differing only in the primer annealing time, which is specific for each pair used: initial denaturation at 94° C for 3min, 35 repeated cycles of 60s at 94° C for denaturation, 60s at X °C for primer annealing (for the 2416T-A mutation: 56°C; IVS8-2A-G: 51°C; 2485C-T: 58°C; 5473C-G mutation: 59°C; 3032T-C mutation: 52°C), and extension of 2minutes at 72°C and 10min at 72°C for the final extension. The fragments amplified by PCR were respectively submitted to RFLP analysis, using 5U of the following enzymes: DdeI enzyme, BGlII, BfaI, FauI and MspI (New England Biolabs®).
The products of the reactions (PCR/RFLP) were added to bromophenol blue and submitted to electrophoresis on 2% agarose gel in Tris–borate–EDTA 1× buffer containing ethidium bromide (0.5ug/mL), after which the gels were illuminated with UV lighting and photographed.
Statistical analysisThe results were previously submitted to descriptive statistics to determine normality. The Chi-squared test was used to compare independent variables with normal distribution and the Kruskal–Wallis test for samples with non-normal distribution, in addition to odds ratio with a confidence interval of 95% (95% CI). The level of significance was set at 5%. Statistical tests were performed using GraphPad InStat software release 3.0 (GraphPad Software Inc – San Diego, CA, United States). Consistency of genotype distribution with the Hardy-Weinberg equilibrium was assessed by exact tests.13
ResultsOf a total of 16 index cases from the NPT group, nine (56%) were females and seven (44%) were males. Age ranged from 8 to 65 years for females (mean M 24.2 years, SD±20.1) and for males between 1 and 38 years (M 14.1 years, SD±15.7), with no statistically significant difference (p=0.29).
Of the 13 patients in the NSHL group, seven (54%) were males and six (46%) were females. Age varied from 7 to 37 years for males (M 22.7 years, SD±12.6) and between 12 and 45 years for females (M 33.3 years, SD±12.3), with no significant difference (p=0.15).
Regarding the 20 individuals in the CG, 13 (65%) were males and seven (35%) were females. Age ranged from 19 to 44 years for males (M 28.5 years, SD±7.2) and between 20–35 years for females (M 26.3 years, SD±6.3), with no significant difference (p=0.49).
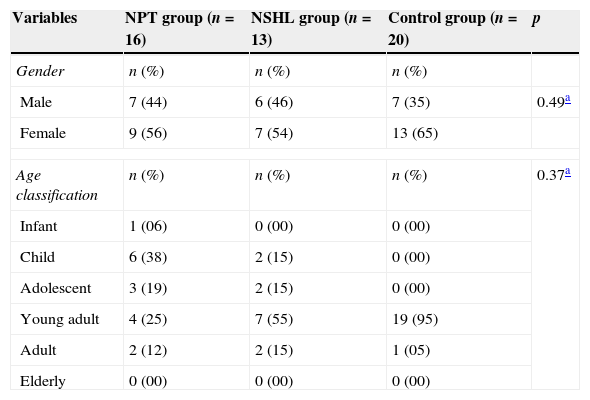
Considering the demographic variables among patients in the three groups, there was a prevalence of females in all three, although it was not significant (p=0.49). Both the NSHL and CG groups had a higher prevalence in the young adult age range, totaling 55% and 95%, respectively. The NPT group had higher prevalence in the infant group (38%). The statistical analysis for this demographic variable was not significant between groups (p=0.37). Similarly, the mean ages among the three groups showed no significant differences (p=0.091; Table 1).
Distribution of demographic variables of index cases in the NPT group, patients from the NSHL group, and individuals from the Control group.
| Variables | NPT group (n=16) | NSHL group (n=13) | Control group (n=20) | p |
|---|---|---|---|---|
| Gender | n (%) | n (%) | n (%) | |
| Male | 7 (44) | 6 (46) | 7 (35) | 0.49a |
| Female | 9 (56) | 7 (54) | 13 (65) | |
| Age classification | n (%) | n (%) | n (%) | 0.37a |
| Infant | 1 (06) | 0 (00) | 0 (00) | |
| Child | 6 (38) | 2 (15) | 0 (00) | |
| Adolescent | 3 (19) | 2 (15) | 0 (00) | |
| Young adult | 4 (25) | 7 (55) | 19 (95) | |
| Adult | 2 (12) | 2 (15) | 1 (05) | |
| Elderly | 0 (00) | 0 (00) | 0 (00) | |
| Age | M±SD | M±SD | M±SD | p |
|---|---|---|---|---|
| Years | 19.8±18.5 | 27.6±13.1 | 19.±18.5 | 0.091b |
M±SD, mean±standard deviation.
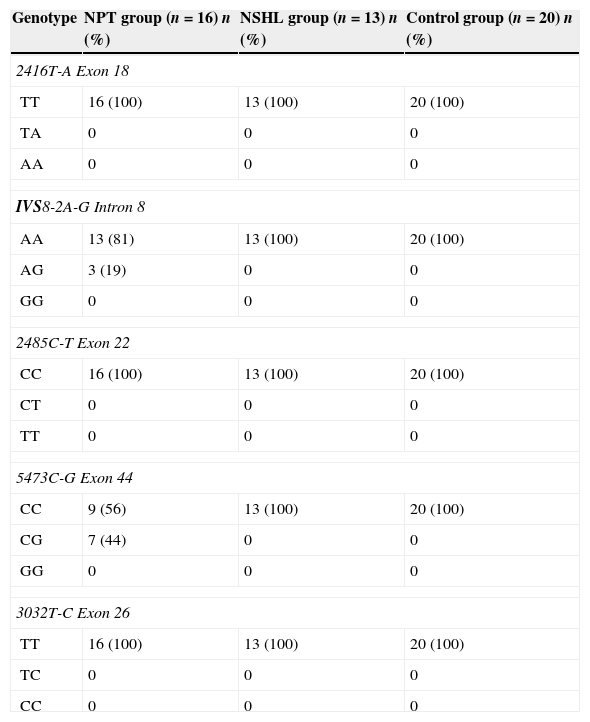
Table 2 shows the distribution, in percentages, of the genotypes found in the three groups assessed for the five mutations in the OTOF gene. In the NPT group, 13 (81%) index cases had the wild genotype (AA) and three (19%) had the heterozygous genotype AG for IVS8-2A-G mutation (intron 8). The 5473C-G mutation (exon 44) was found in heterozygous form (CG) in seven (44%) of the index cases and nine (56%) had the wild allele (CC). In the NSHL and CG groups, 100% of patients had none of the analyzed mutations in the OTOF gene.
Distribution, in percentages, of the genotypes found in the three groups assessed for the five mutations in the OTOF gene.
| Genotype | NPT group (n=16) n (%) | NSHL group (n=13) n (%) | Control group (n=20) n (%) |
|---|---|---|---|
| 2416T-A Exon 18 | |||
| TT | 16 (100) | 13 (100) | 20 (100) |
| TA | 0 | 0 | 0 |
| AA | 0 | 0 | 0 |
| IVS8-2A-G Intron 8 | |||
| AA | 13 (81) | 13 (100) | 20 (100) |
| AG | 3 (19) | 0 | 0 |
| GG | 0 | 0 | 0 |
| 2485C-T Exon 22 | |||
| CC | 16 (100) | 13 (100) | 20 (100) |
| CT | 0 | 0 | 0 |
| TT | 0 | 0 | 0 |
| 5473C-G Exon 44 | |||
| CC | 9 (56) | 13 (100) | 20 (100) |
| CG | 7 (44) | 0 | 0 |
| GG | 0 | 0 | 0 |
| 3032T-C Exon 26 | |||
| TT | 16 (100) | 13 (100) | 20 (100) |
| TC | 0 | 0 | 0 |
| CC | 0 | 0 | 0 |
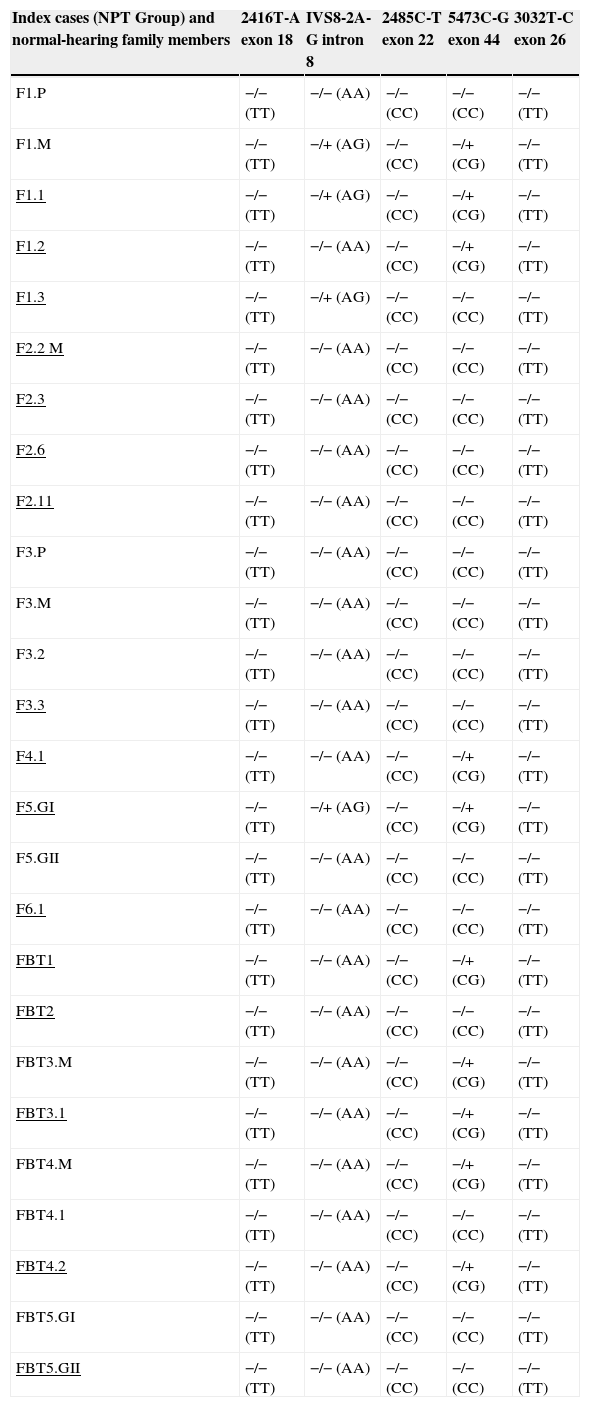
Thus, in the NPT group, there were eight (50%) index cases with mutations in the OTOF gene, one (12.5%; F1.3) heterozygous for the mutation IVS8-2A-G, five (62.5%; F1.2, F4.1, FBT1, FBT3.2, and FBT4.2) compound heterozygous for the mutation 5473C-G, and two (25%; F1.1 and F5GI) compound heterozygous for the mutations found respectively in intron 8 and exon 44. Of the normal-hearing family members, the mother of Family 1 (F1.M) is a compound heterozygous for the mutations analyzed in intron 8 and exon 44 and the mothers of Families FBT3 and FBT4 are heterozygous only for the 5473C-G mutation (Table 3).
Genotypes found in the NPT group index cases among the five mutations assessed in the OTOF gene.
| Index cases (NPT Group) and normal-hearing family members | 2416T-A exon 18 | IVS8-2A-G intron 8 | 2485C-T exon 22 | 5473C-G exon 44 | 3032T-C exon 26 |
|---|---|---|---|---|---|
| F1.P | −/− (TT) | −/− (AA) | −/− (CC) | −/− (CC) | −/− (TT) |
| F1.M | −/− (TT) | −/+ (AG) | −/− (CC) | −/+ (CG) | −/− (TT) |
| F1.1 | −/− (TT) | −/+ (AG) | −/− (CC) | −/+ (CG) | −/− (TT) |
| F1.2 | −/− (TT) | −/− (AA) | −/− (CC) | −/+ (CG) | −/− (TT) |
| F1.3 | −/− (TT) | −/+ (AG) | −/− (CC) | −/− (CC) | −/− (TT) |
| F2.2 M | −/− (TT) | −/− (AA) | −/− (CC) | −/− (CC) | −/− (TT) |
| F2.3 | −/− (TT) | −/− (AA) | −/− (CC) | −/− (CC) | −/− (TT) |
| F2.6 | −/− (TT) | −/− (AA) | −/− (CC) | −/− (CC) | −/− (TT) |
| F2.11 | −/− (TT) | −/− (AA) | −/− (CC) | −/− (CC) | −/− (TT) |
| F3.P | −/− (TT) | −/− (AA) | −/− (CC) | −/− (CC) | −/− (TT) |
| F3.M | −/− (TT) | −/− (AA) | −/− (CC) | −/− (CC) | −/− (TT) |
| F3.2 | −/− (TT) | −/− (AA) | −/− (CC) | −/− (CC) | −/− (TT) |
| F3.3 | −/− (TT) | −/− (AA) | −/− (CC) | −/− (CC) | −/− (TT) |
| F4.1 | −/− (TT) | −/− (AA) | −/− (CC) | −/+ (CG) | −/− (TT) |
| F5.GI | −/− (TT) | −/+ (AG) | −/− (CC) | −/+ (CG) | −/− (TT) |
| F5.GII | −/− (TT) | −/− (AA) | −/− (CC) | −/− (CC) | −/− (TT) |
| F6.1 | −/− (TT) | −/− (AA) | −/− (CC) | −/− (CC) | −/− (TT) |
| FBT1 | −/− (TT) | −/− (AA) | −/− (CC) | −/+ (CG) | −/− (TT) |
| FBT2 | −/− (TT) | −/− (AA) | −/− (CC) | −/− (CC) | −/− (TT) |
| FBT3.M | −/− (TT) | −/− (AA) | −/− (CC) | −/+ (CG) | −/− (TT) |
| FBT3.1 | −/− (TT) | −/− (AA) | −/− (CC) | −/+ (CG) | −/− (TT) |
| FBT4.M | −/− (TT) | −/− (AA) | −/− (CC) | −/+ (CG) | −/− (TT) |
| FBT4.1 | −/− (TT) | −/− (AA) | −/− (CC) | −/− (CC) | −/− (TT) |
| FBT4.2 | −/− (TT) | −/− (AA) | −/− (CC) | −/+ (CG) | −/− (TT) |
| FBT5.GI | −/− (TT) | −/− (AA) | −/− (CC) | −/− (CC) | −/− (TT) |
| FBT5.GII | −/− (TT) | −/− (AA) | −/− (CC) | −/− (CC) | −/− (TT) |
Underlined, index cases with auditory neuropathy (NPT). F, family; P, father of the index case; M, mother of the index case; GI, twin; I GII, twin II; −/−, wild homozygous genotype for the analyzed mutation; −/+, heterozygous genotype for the analyzed mutation; −/+, heterozygous genotype for the mutation analyzed in normal-hearing family member.
The NPT group was assessed for the Hardy–Weinberg equilibrium in relation to mutations found in intron 8 and exon 44. According to the calculations made, allelic frequencies A (wild-type allele) and G (mutant allele) of the IVS8-2A-G mutation, for the index cases of the NPT group, were A=0.91 and G=0.09. The observed genotypic frequencies of the wild homozygous form (AA-13), mutant heterozygous (AG-3), and mutant homozygous (GG-0) of these index cases are consistent with the expected frequencies for the Hardy-Weinberg equilibrium (χ2=0.171; p=0.68). Regarding the 5473C-G mutation, the allelic frequencies C (wild-type allele) and G (mutant allele) for the index cases of the NPT group were C=0.78 and G=0.22. For this mutation, the genotypic frequencies observed in the wild homozygous form (CC-9), mutant heterozygous (GC-7), and mutant homozygous (GG-0) of these index cases are also in agreement with the expected frequencies for the Hardy–Weinberg equilibrium (χ2=0.125; p=0.26).
Regarding the degree of hearing loss found in the index cases of the NPT group, three (19%) had mild, five (31%) moderate, five (31%) severe, and three (19%) profound hearing loss. In the NSHL group, four (31%) had moderate, two (15%) severe, and seven (54%) profound hearing loss. There were no mild cases in this group. Statistical analysis for the variable degree of hearing loss showed no significant difference between groups (p=0.124).
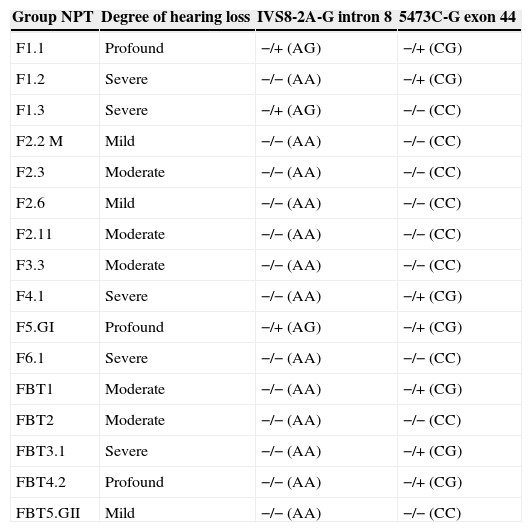
Due to mutation identification only in intron 8 and exon 44 in the OTOF gene, Table 4 shows the degree of hearing loss in the NPT group index cases and genotypes found in these cases, through the molecular analysis of these regions.
Degree of hearing loss and the genotypes found in Intron 8 and Exon 44 of the OTOF gene, in index cases of NPT group.
| Group NPT | Degree of hearing loss | IVS8-2A-G intron 8 | 5473C-G exon 44 |
|---|---|---|---|
| F1.1 | Profound | −/+ (AG) | −/+ (CG) |
| F1.2 | Severe | −/− (AA) | −/+ (CG) |
| F1.3 | Severe | −/+ (AG) | −/− (CC) |
| F2.2 M | Mild | −/− (AA) | −/− (CC) |
| F2.3 | Moderate | −/− (AA) | −/− (CC) |
| F2.6 | Mild | −/− (AA) | −/− (CC) |
| F2.11 | Moderate | −/− (AA) | −/− (CC) |
| F3.3 | Moderate | −/− (AA) | −/− (CC) |
| F4.1 | Severe | −/− (AA) | −/+ (CG) |
| F5.GI | Profound | −/+ (AG) | −/+ (CG) |
| F6.1 | Severe | −/− (AA) | −/− (CC) |
| FBT1 | Moderate | −/− (AA) | −/+ (CG) |
| FBT2 | Moderate | −/− (AA) | −/− (CC) |
| FBT3.1 | Severe | −/− (AA) | −/+ (CG) |
| FBT4.2 | Profound | −/− (AA) | −/+ (CG) |
| FBT5.GII | Mild | −/− (AA) | −/− (CC) |
Among the index cases with mutation, those with the severe (four; 25%) and profound (three; 19%) hearing loss were more prevalent than those without mutation (only one severe HL, 6%) of the same group, which showed a higher prevalence of mild (three; 19%) and moderate (four; 25%) HL, with a significant difference (p=0.022).
DiscussionAN is a hearing disorder characterized by severe distortion or absence of BAEP with preserved OAEs, thus indicating the presence of outer hair cell integrity.14
Among the previously identified causes of AN are hyperbilirubinemia, neonatal hypoxia, or neurodegenerative diseases. However, there have been reports of “isolated” AN, i.e., without any risk factors, and no physiopathological mechanism has been proposed to explain these cases. Recent advances in genetic research has allowed a new approach to elucidate the physiopathology of this disease.15,16
Given the above, this study aimed to investigate the prevalence of five mutations in the OTOF gene in patients with AN diagnosis, in normal-hearing subjects and in patients with NSHL. These were included in order to determine whether mutations in the OTOF gene could be identified in normal-hearing subjects and in patients with NSHL without mutations in other genes and without AN.
Regarding the demographic variables, there was a prevalence of females in the three study groups, although it was not statistically significant. The NSHL and CG groups had a higher prevalence in the young adult age range and the NPT group had a higher prevalence in the infant age range, but without a significant difference. These data are consistent with the few published reports on the prevalence of gender and age range related to the OTOF gene.17,18
Most reports of mutations identified in the OTOF gene have been described in homozygous form and in consanguineous families.3,4,6,9,18–22 Among the five mutations analyzed in the OTOF gene, only the IVS8-2A-G mutation in intron 8 and 5473C-G mutation in exon 44 were found, affecting 50% of the study index cases. In contrast to the literature, all index cases were the offspring of non-consanguineous marriages, demonstrating that mutations in the OTOF gene that are associated with AN may occur in these cases.
Heterozygous cases in patients with AN have been described, with or without consanguinity.2,5,8,10–12,23,24 In the present study, the IVS8-2A-G mutation was found in heterozygous state (one mutant allele) in 12.5% of index cases, with the same occurring with the 5473C-G mutation, which was found in 62.5% of cases. In 25% of cases, the two mutations identified occurred at the same time in the same individual, which, therefore, are called compound heterozygotes. These two mutations, as well as the other three analyzed, were not found in the NSHL group (100%) nor in the CG group. These data are in agreement with the literature.2,5,8,10–12,23,24
In 2000, the IVS8-2A-G mutation was first described in its homozygous state in three brothers from India born to consanguineous parents and in its heterozygous state in the parents and unaffected siblings, and was not identified in 109 normal-hearing individuals from the control group.6 There have been no further reports on this mutation. The 5473C-G mutation was described in compound heterozygosity in a Spanish family, with the 2485C-T mutation in exon 22, identified in another allele.5
Similarly, there have been no further reports of the aforementioned mutation in exon 44.
The IVS8-2A-G mutation, by creating a premature stop codon, and the 5473C-G mutation, by altering the binding capacity with Ca+2 in the wild-type gene, are considered pathogenic when in their homozygous state.3,5 In this study, both were found in compound heterozygous states only in index cases with AN (F1.1 and F5.GI), which could explain the molecular cause of hearing loss in these individuals, as the AN phenotype has been observed in patients with two mutations affecting all isoforms of otoferlin or one mutation affecting the long isoform and another, the short isoform. This means that any combination of biallelic mutations in the OTOF gene may result in AN, as they are partially related to the nature (truncated or non-truncated) or the location (long or short isoform) of these mutations.23
However, the same mutations were also found in compound heterozygosis in the normal-hearing mother (F1.M) of the compound heterozygous index case (F1.1). As the mother has normal hearing, the cause of HL in the index case may be due to other existing mutations in other exons or other splice sites, or more complex mutations, such as large deletions or other sequence rearrangements, which require a more specific molecular approach than used here. Similarly, these hypotheses may also be applied to explain the other six HL index cases, as they are carriers of one of the mutations identified in only one of the alleles.
Brazilian references have not reported the mutations identified in this study,10–12 and only one of them identified the 2485C-T mutation in exon 22 with heterozygosity in 0.5% of cases of patients with non-syndromic sensorineural HL (1/200).10
Estimates of the prevalence of AN in deaf individuals range from 1% to 19%. 25–28 In Brazil, the prevalence of AN among all cases of deafness is 1.2%.29 This range of variation may be due to the fact that different populations were studied or due to differences in the criteria used to identify patients with this disease. The prevalence of AN in a population without risk factors is not well established.9 In the present study, all the criteria for inclusion and exclusion were strictly followed, thus determining the prevalence of 1.3% of cases with AN in a population with sensorineural hearing loss during the study period.
Moreover, the Brazilian estimate of the magnitude of HL in patients with AN is 29.6% mild, 55.5% moderate, 7.4% severe, and 7.5% profound HL.29 In the present study 19% had mild, 31% moderate, 31% severe, and 19% profound HL. Among the index cases with mutations in the OTOF gene, we found a statistically significant prevalence of severe and profound HL compared to the mild and moderate HL in index cases without mutation. The intensity of hearing loss caused by AN can range from mild to profound, with a disproportionate decrease in understanding speech that does not correspond to the pure-tone audiogram.
There is some homogeneity in the description of the phenotype of patients with mutations in the OTOF gene, that is characterized by a congenital or pre-lingual profound HL not associated with other abnormalities, as the index cases studied. Therefore, AN is the only phenotypic expression of mutations in the OTOF gene, with hearing loss identified by the relative preservation of outer hair cell function.9,30
The presence of OAEs in the first months of life has important implications for neonatal screening programs, such as initial screening. In the case of AN, there is a risk that this loss will go undiagnosed, for although these patients have severe to profound degree of hearing loss, they would be included among the cases classified as “no alteration or normal” because OAEs reflect the auditory function in outer hair cells and their presence does not exclude alterations of inner hair cells, in synapses, or in the auditory nerve.28
In the context of HL investigation, it is important to take into account the type of AN loss, which is often misdiagnosed. Therefore, when assessing a child or patient with deafness in the presence of OAEs, the hypothesis of AN has to be raised and genetic testing for mutations in the OTOF gene research should be performed, as they were performed in this study, allowing an accurate etiologic diagnosis to provide appropriate audiological treatment in a timely manner.28
Potential limitations regarding the study findings deserve consideration; despite the positive results, they should be interpreted with caution and should be corroborated by independent, multicenter studies to determine the true prevalence of mutations in the OTOF gene and the association with AN in the Brazilian population.
ConclusionThe evidence of association between mutations in the OTOF gene and AN indicates that there are differences at the molecular level in patients with and without this type of hearing loss; in the absence of risk factors, the combination of severe distortion or absence of BAEP in the presence of OAEs should direct the diagnosis of AN and the screening for mutations in the OTOF gene. Molecular tests are valuable diagnostic complements, not only individually, but also as a screening method for a neonatal or population study.
FundingThis study was supported by Research Grant (BAP-FAMERP).
Conflicts of interestThe authors declare no conflicts of interest.
To the patients and their parents/guardians who, by agreeing to participate in this research, will contribute to a better future for the children of Brazil. To the institution in which this research was performed, by encouraging research and teaching qualification.
Please cite this article as: da Silva MA, Piatto VB, Maniglia JV. Molecular approach of auditory neuropathy. Braz J Otorhinolaryngol. 2015;81:321–8.


