




A etiologia da perda auditiva depende da população estudada, da etnia e da condição socioeconômica da região analisada. O diagnóstico etiológico contribui para o aprimoramento das medidas preventivas e para a identificação precoce dessa deficiência.
ObjetivosIdentificar os fatores etiológicos da perda auditiva e sua prevalência em um hospital terciário do sul do Brasil, verificar a frequência de mutações nos genes GJB2 e GJB6 e correlacionar o grau da perda auditiva com os fatores etiológicos da deficiência auditiva.
MétodoEste estudo de prevalência avaliou 140 crianças com perda auditiva neurossensorial bilateral ou mista. Foram submetidos a anamnese com histórico médico, exame físico, audiometria e potencial evocado auditivo de tronco encefálico. Exames de imagem e genéticos também foram feitos.
ResultadosAs etiologias e sua prevalência foram as seguintes: (a) causas indeterminadas, 31,4%; (b) condições relacionadas ao período neonatal, 22,1%; (c) genética, 22,1%; (d) neuropatia auditiva, 10%; (e) outros fatores (malformação cortical, hemorragia intracraniana e malformações da orelha interna), 7,9% e (f) infecções congênitas, 6,4%. Entre os casos genéticos, foram identificados dez casos homozigotos e sete heterozigotos da mutação 35delG, além de dois casos de variantes raras do GJB2: p.Try172* e p.Arg184Pro. Foi encontrado um caso homozigoto da mutação del (GJB6‐D13S1830). Em relação à gravidade da perda auditiva, em 78,6% dos casos o grau da perda auditiva foi profundo e não houve diferenças significantes na comparação entre as etiologias.
ConclusãoO número de etiologias indeterminadas ainda é elevado e a infecção congênita por CMV pode ser uma possível causa de etiologia não diagnosticada para perda auditiva. A predominância das etiologias relacionadas às condições neonatais e às causas infecciosas são características de países em desenvolvimento. A mutação mais prevalente foi a 35delG e o principal gene foi o GJB2, provavelmente devido à influência europeia no genótipo de nossa população.
A deficiência auditiva é a deficiência sensorial congênita mais comum, com uma incidência de um a dois casos por 1.000 nascidos vivos.1–7 Essa prevalência significativa resulta em um grande impacto sociocultural, uma vez que a deficiência auditiva interfere significativamente nos processos de apropriação da linguagem oral e escrita. A limitação do acesso à oralidade, por si só, requer inúmeras adaptações nas diversas relações sociais e familiares.5,7–9
Os principais fatores etiológicos da perda auditiva têm prevalência variável, pois são diretamente influenciados pelo desenvolvimento socioeconômico, etnia e região de um determinado país.3 Em países desenvolvidos, a maioria dos casos de perda auditiva é de natureza genética (50%‐60% dos casos), que compreende as formas sindrômicas (15%‐30%) e as formas não sindrômicas (70%‐85%).10 Do restante, aproximadamente 35% estão relacionados a doenças infecciosas ou secundárias a eventos neonatais.11 A realidade é diferente nos países em desenvolvimento, onde, apesar das melhorias no cuidado neonatal e programas de imunização, condições relacionadas ao período neonatal e infecções ainda são causas muito comuns de perda auditiva.12
A avaliação diagnóstica da perda auditiva infantil inclui a investigação da história do paciente, exames físico e audiológico e testes eletrofisiológicos. Ao fazer investigações adicionais, como avaliação multidisciplinar, testes genéticos e de imagem,8 é possível identificar a etiologia em 50% a 60% de todos os casos de perda auditiva.12
Os testes genéticos mais importantes são aqueles que investigam as mutações dos genes Gap Junction Protein Beta 2 (GJB2) e Gap Junction Protein Beta 6 (GJB6), que codificam as conexinas 26 e 30, respectivamente. As mutações GJB2 são as mais prevalentes, são responsáveis por mais de 50% dos casos de perda auditiva por causas genéticas não sindrômica. A mutação 35delG do gene GJB2 é a mais comum.1 Em casos com heterozigose no gene GJB2, onde não há uma segunda mutação no mesmo gene que justifique o fenótipo da deficiência auditiva, a investigação do GJB6 é recomendada, uma vez que grandes deleções no GJB6 geralmente ocorrem em combinação com mutações recessivas no GJB2.13,14 As principais mutações do GJB6 são del(GJB6‐D13S1830) e del(GJB6‐D13S1854).13
A identificação das principais etiologias da deficiência auditiva infantil é importante para a prevenção primária dessa deficiência, por meio de medidas de saúde pública como vacinação e melhora da saúde materno‐infantil. O diagnóstico precoce da deficiência auditiva genética também pode contribuir para medidas preventivas, através do aconselhamento genético.5,15 Além disso, a miscigenação da população brasileira,6 com diferentes etnias nas diversas regiões brasileiras, pode indicar a existência de mutações genéticas distintas entre elas. Essa realidade torna importante investigar a genética da perda auditiva em cada região.
O principal objetivo deste estudo foi identificar as etiologias mais prevalentes de perda auditiva em um hospital terciário do sul do Brasil. Os objetivos secundários foram verificar a prevalência de mutações nos genes das conexinas 26 e 30 e correlacionar o grau da perda auditiva com a sua etiologia.
MétodoLocais básicos do estudo e seleção de pacientesEsta é uma coorte retrospectiva na qual foram incluídos pacientes atendidos em um ambulatório de um centro de referência em deficiência auditiva infantil de um hospital terciário do sul do Brasil. Os encaminhamentos para essa clínica ocorreram por dois motivos: (a) falha na triagem auditiva neonatal (TAN) ou (b) investigação e tratamento de perda auditiva já diagnosticada.
Os critérios de inclusão para este estudo foram os seguintes: (a) até 12 anos incompletos; (b) perda auditiva neurossensorial bilateral ou mista; e (c) perda auditiva congênita ou adquirida no período neonatal. O projeto foi aprovado pelo comitê de ética em pesquisa do hospital onde a pesquisa foi feita (número 150464). Todos os adultos responsáveis pelas crianças assinaram o termo de consentimento livre e esclarecido.
Avaliação dos pacientesNa primeira consulta de cada paciente, foi coletada uma história médica para obter informações importantes a fim de caracterizar o perfil da criança, a presença de fatores de risco para deficiência auditiva e obter hipóteses para o diagnóstico etiológico dessa deficiência. Os fatores de risco avaliados para perda auditiva na infância foram os definidos pelo Joint Committee on Infant Hearing.16
As perdas auditivas adquiridas antes do nascimento, independentemente da época de manifestação dos sintomas, foram consideradas como congênitas neste estudo. O termo “condições relacionadas ao período neonatal” foi usado para descrever condições ou complicações neonatais que causam perda auditiva e ocorrem logo após o nascimento e podem se estender além do primeiro mês de vida do recém‐nascido. Essas condições/ complicações são parte dos fatores de risco determinados pelo Joint Committee on Infant Hearing, 2007.16
Para demonstrar que a perda auditiva era congênita ou adquirida no período neonatal, o primeiro critério foi a etiologia da deficiência auditiva. Em caso de etiologia indeterminada, consideramos o resultado da TAN. Assim, partimos do pressuposto de que pacientes com perda auditiva sem etiologia definida que apresentaram falha na TAN tinham etiologia congênita.
Avaliação audiológicaO tipo de perda auditiva foi definido pelo potencial evocado auditivo em frequência específica e/ou audiometria de reforço visual (lúdica/comportamental ou tonal e vocal, de acordo com a idade e habilidade da criança em responder ao exame). A avaliação do grau de perda auditiva foi feita através do Interacoustics Eclipse EP25 ABR system® (Dinamarca) com NB‐chirps® em 500Hz, 1000Hz, 2000Hz e 4000Hz ou com o audiômetro Interacoustics AD 27 (com ou sem fone supra‐aural TDH‐39) e classificado de acordo com a classificação da Organização Mundial da Saúde (OMS, 2014),17 que usa a média quadrática entre os limiares auditivos para as frequências de 500Hz, 1000Hz, 2000Hz e 4000Hz. Essa classificação divide os limiares auditivos em leve (entre 26 e 30dB), moderado (entre 31 e 60dB), grave (entre 61 e 80dB) e profundo (maior que 81dB). Pacientes com otite média com efusão foram submetidos à timpanotomia para colocação de tubo de ventilação antes da classificação definitiva do tipo e grau da perda auditiva.
Exames laboratoriais e de imagemOs exames laboratoriais para investigação das infecções congênitas foram feitos durante o pré‐natal das gestantes. Nos casos em que a mãe não havia feito o pré‐natal, a investigação foi feita no momento do parto e, a depender do resultado, também no recém‐nascido, através de testes sorológicos, conforme recomendação do Ministério da Saúde do Brasil.18
A sorologia para citomegalovírus (CMV) também não foi investigada, devido à baixa sensibilidade e especificidade da presença da imunoglobulina M (IgM) no diagnóstico da infecção congênita por esse vírus. O isolamento do vírus em cultura de fibroblastos humanos ou detecção de DNA viral por reação em cadeia da polimerase (PCR) foram feitos em amostras de urina ou saliva dos bebês. Esse teste foi feito apenas em bebês sintomáticos.19
Pacientes com suspeita de malformação da orelha interna (anomalias ou síndromes craniofaciais, por exemplo) ou candidatos ao implante coclear com perda auditiva grave ou profunda foram submetidos a exames de imagem: tomografia computadorizada (TC) e/ou ressonância magnética (RM) de ossos temporais. Pacientes com suspeita de síndromes, distúrbios neurológicos ou oftalmológicos foram encaminhados para avaliação especializada.
Testes genéticosAmostras de sangue foram coletadas de todas as crianças não sindrômicas para extração de DNA e análise de mutações do gene GJB2 e da deleção del(GJB6‐D13S1830) do gene GJB6. Cinco mL de sangue foram coletados em tubo com ácido rtilenodiaminotetracético (EDTA) e o material foi enviado ao laboratório de genética médica para extração de ácido desoxirribonucleico (DNA) com o Kit GENTRA Puregene (Qiagen Inc., Valencia, EUA).
Para a análise genética da conexina 30, as amostras foram submetidas a PCR para isolamento do fragmento de interesse, com três primers (1 forward e 2 reversos).20 O uso de três primers na mesma reação permite avaliar a presença da deleção del(GJB6‐D13S1830) no gene GJB6, bem como se ela está presente em heterozigose ou homozigose. Os fragmentos foram visualizados por eletroforese em gel de agarose a 2%. Para a análise genética da conexina 26, as amostras também foram submetidas a PCR para amplificação. Em seguida, os materiais genéticos amplificados foram preparados e submetidos ao sequenciamento bidirecional pelo método de Sanger.21
Grupos etiológicosPara facilitar a comparação dos dados encontrados com os de outros estudos, agrupamos os fatores etiológicos em seis grupos principais:
- 1)
Condições relacionadas ao período neonatal (CRPN): internação em unidade de terapia intensiva neonatal (UTIN) > 5 dias ou ventilação extracorpórea, ventilação assistida, uso de medicamentos ototóxicos (gentamicina e tobramicina) ou diuréticos de alça (furosemida) e hiperbilirrubinemia com necessidade de transfusão.16 Todos esses fatores foram classificados em uma única categoria para fins de análise e porque os pacientes geralmente apresentam vários deles ao mesmo tempo. Os pacientes foram incluídos neste grupo após a exclusão dos seguintes fatores etiológicos: infecção congênita e neonatal, distúrbios genéticos sindrômicos e não sindrômicos, malformação da orelha interna e distúrbios do espectro da neuropatia auditiva.
- 2)
Infecção congênita ou neonatal (ICN): infecções adquiridas no período pré e perinatal, como sífilis, toxoplasmose, rubéola, CMV, herpes vírus e vírus da imunodeficiência humana adquirida (HIV).
- 3)
Genética: perda auditiva sindrômica (PAS) e perda auditiva não sindrômica (PANS). A PAS consiste em PA que se apresenta juntamente com anomalias visuais, renais, do sistema musculoesquelético e nervoso, bem como distúrbios pigmentares ou outros, ao passo que, em nosso estudo, PANS refere‐se a pacientes com mutação genética no gene GJB2 e/ou GJB6 comprovada por exame molecular.
- 4)
Transtorno do espectro da neuropatia auditiva (TENA): definido como a presença de emissões otoacústicas e/ou microfonismo coclear e a ausência de respostas ou alterações significativas no PEATE. 22,23
- 5)
Indeterminado: após excluir outras causas de perda auditiva;
- 6)
Outros: malformações da orelha externa, média ou interna e etiologias centrais.
Outro tipo de classificação que fizemos foi de acordo com as etapas do processo de investigação da etiologia causadora da perda auditiva. O motivo para isso foi determinar quantos pacientes podem ser diagnosticados em uma ou duas etapas e quantos precisam de procedimentos mais complexos.
Os grupos e critérios usados para definir cada um foram os seguintes:
- 1)
História médica e exame físico: podem identificar condições relacionadas ao período neonatal e infecções congênitas ou neonatais, bem como a maioria das síndromes.
- 2)
Exames auditivos: podem detectar o TENA através de achados sugestivos nas emissões otoacústicas e PEATE.
- 3)
Exames de imagem: podem detectar a presença de malformações na orelha interna;
- 4)
Testes genéticos: identificam a presença de mutações nos genes investigados que confirmam se a perda auditiva tem origem genética não sindrômica;
- 5)
Avaliação por especialista: quando pacientes com um conjunto de alterações sugestivas de síndromes são encaminhados para avaliação no serviço de genética médica para o diagnóstico etiológico.
Foi feita uma análise estatística descritiva. As variáveis quantitativas foram descritas por média ± desvio‐padrão e as variáveis categóricas, por frequência total e absoluta. O teste de qui‐quadrado de Pearson foi usado para comparar as variáveis categóricas. O software SPSS 18.0 foi usado na análise estatística (SPSS Inc. lançado em 2009. PASW Statistics for Windows, versão 18.0, Chicago: SPSS Inc).
ResultadosNo período de janeiro de 2015 a dezembro de 2017, 258 crianças foram encaminhadas ao ambulatório: 103 pacientes (39,9%) por apresentarem falha na TAN e 155 pacientes (60,1%) para investigação e tratamento de perda auditiva já diagnosticada. A deficiência auditiva foi confirmada em 239 (92,6%) pacientes, foi bilateral em 213 (82,5%). Desses, 140 (54,2%) pacientes atenderam aos critérios de inclusão, como apresentado no organograma da figura 1.

Após a análise inicial, foram avaliadas 140 crianças com média de idade (± desvio‐padrão) na primeira consulta de 2,00 anos (± 2,29 anos, 1 a 12 anos). Setenta e dois pacientes (51,4%) eram do sexo masculino. Quanto à etnia/cor da pele, 89,3% se identificaram como brancos, 7,9% negros, 2,1% pardos e 0,7% indígenas. Sessenta e oito crianças (50,7%) eram filhos de mães multíparas e 98,5% das mães fizeram consultas de pré‐natal. Em relação ao grau da perda auditiva na melhor orelha, a média foi de 96,43dB (± 25,05).
Entre as crianças avaliadas, 85 (60,7%) tinham pelo menos um fator de risco para perda auditiva na infância.16 Vinte e sete crianças (19,3%) tinham história familiar de perda auditiva – nove tinham um(a) irmão(ã) e oito tinham um dos pais com deficiência auditiva. Não houve casos relatados de consanguinidade.
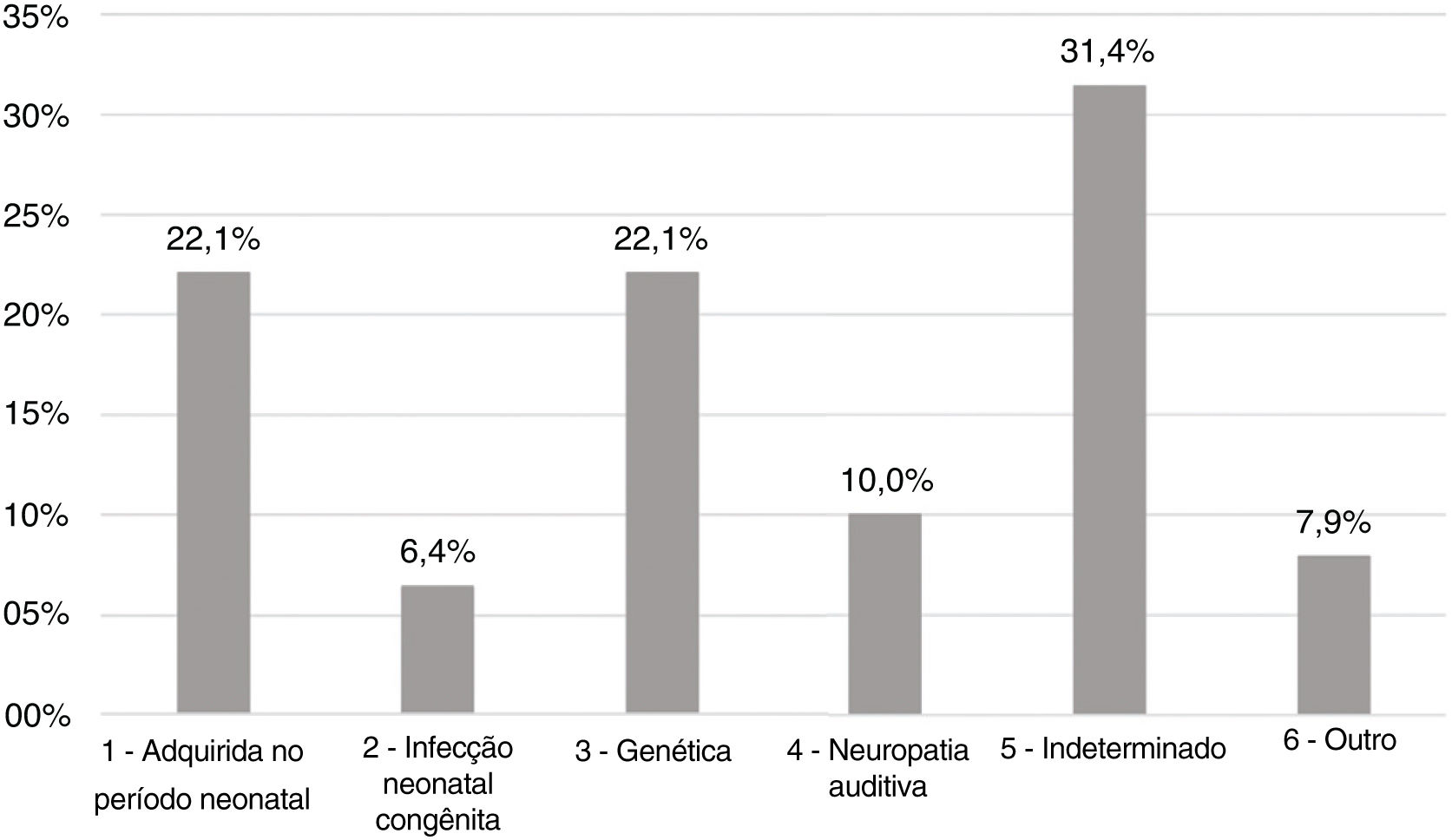
Achados etiológicosOs grupos etiológicos mais prevalentes foram os de condições relacionadas ao período neonatal (Grupo 1) e à genética (Grupo 3), com 31 casos (22,1%) cada. Em 44 pacientes (31,4%) não foi possível determinar a causa do déficit auditivo (Grupo 5), como demonstrado na figura 2.

No Grupo 1 (CRPN ‐ condições relacionadas ao período neonatal), a permanência na UTI neonatal por mais de cinco dias foi o fator mais prevalente associado à perda auditiva, ocorreu em 48 pacientes (34,3%), seguida de ventilação mecânica (20,7%), uso de ototóxicos (19,3%) e transfusão de troca sanguínea para icterícia neonatal (6,4%).
A infecção por CMV foi a mais prevalente do Grupo 2, com quatro casos (2,9%). Também foram identificados dois casos (1,4%) de sífilis congênita, 2 (1,4%) de toxoplasmose e um (0,7%) de infecção pelo herpes vírus. Todas essas crianças foram diagnosticadas com infecção congênita antes da primeira consulta no ambulatório. Sete crianças eram sintomáticas: quatro com infecção por CMV, duas com toxoplasmose e uma com herpes vírus.
A perda auditiva de origem genética foi dividida em sindrômica, 41,93%, e não sindrômica, 58,06%. Entre os casos sindrômicos, encontramos dois casos de síndrome Charge, um de síndrome de Goldenhar, um de síndrome de Waardenburg, um de síndrome de Treacher‐Collins, um de síndrome de Stickler e sete casos de perda auditiva sindrômica ainda sem diagnóstico definitivo.
Os genótipos dos 18 pacientes com perda auditiva genética não sindrômica e o número de casos de cada um são apresentados na tabela 1.
Genótipo da perda auditiva genética não sindrômica
| Genótipos | n |
|---|---|
| 35delG/35delG | 10 |
| 35delG/N | 5 |
| 35deG/ p. Try172* | 1 |
| 35delG/p.Arg184Pro | 1 |
| del(GJB6‐ D13S1830)/ del(GJB6‐ D13S1830) | 1 |
| N/N | 113 |
35delG/35delG, Homozigoto para 35delG; 35delG/N, Heterozigoto para 35delG; 35deG/p.Try172* e 35delG/p.Arg184Pro, Heterozigoto para 35delG com variantes patogênicas de GJB2; N/N, sem mutações.
Embora os testes genéticos tenham sido feitos em todas as crianças não sindrômicas, independentemente de qualquer outra etiologia possível para a perda auditiva, apenas aquelas sem outra etiologia hipotética apresentaram testes genéticos alterados. Um achado importante foi que a etiologia final em 44,44% das crianças com história familiar positiva de perda auditiva era genética. Desses casos, 29,6% eram não sindrômicos (seis casos homozigotos e dois heterozigotos para a mutação 35delG) e 14,8% eram sindrômicos (um com síndrome de Charge, um com síndrome de Waardenburg, um caso de síndrome de Treacher‐Collins e um em investigação, associado à paralisia do nervo facial periférico).
Exames de imagem (TC e/ou RM de ossos temporais) foram feitos em 131 crianças (93,6%). Oito dessas crianças (6,1%) apresentavam malformações de orelha interna. Entre essas, encontramos um caso de cavidade vestibulococlear única, uma de partição incompleta tipo 1, três de partição incompleta tipo 2, um de aqueduto vestibular dilatado e dois de agenesia do nervo coclear. Entre as malformações da orelha externa e média: três casos de microtia e agenesia de meato acústico externo bilateral, um deles era associado à malformação de orelha média. Todas as malformações da orelha externa foram encontradas em pacientes sindrômicos (um caso de Goldenhar, um caso de Treacher Collins e uma síndrome em investigação), ou seja, não foram consideradas como as principais etiologias. Além disso, dos casos classificados como sendo de etiologia do sistema nervoso central, um foi devido à malformação cortical e dois à sequela de hemorragia intracraniana.
Achados das etapas do diagnósticoConsiderando todas as crianças com etiologia estabelecida para a perda auditiva, 49 (51%) tiveram a etiologia estabelecida na primeira consulta, através de história clínica e exame físico. Os testes genéticos definiram a causa em 18,8% dos casos. Na figura 3, encontram‐se todas as etapas do processo de investigação da etiologia causadora da perda auditiva, com o percentual de casos causados pela mesma.
Achados audiológicos
A figura 4 mostra a distribuição em graus de acordo com a classificação da Organização Mundial da Saúde (2014). Em 78,6% dos casos, o grau de perda auditiva foi profundo. Não houve diferença estatisticamente significante na comparação dos graus da perda auditiva com as principais etiologias.
Discussão.")
De acordo com este estudo, as duas etiologias mais prevalentes para a perda auditiva foram a CRPN e a ICN, as quais foram responsáveis por um maior número de casos quando comparadas à etiologia por causas genéticas. Entre os grupos de PANS, a mutação 35delG do gene GJB2 foi responsável pela maioria dos casos.
É difícil comparar nossos resultados com os de outros estudos. As diferenças de métodos entre o nosso e outros estudos estão principalmente nos critérios de inclusão (lateralidade, tipos e graus da perda auditiva) e nas classificações (dos fatores de risco, categorias de etiologia, graus da perda auditiva). Porém, por se tratar de um estudo de prevalência, espera‐se que os resultados variem de acordo com as características genéticas e socioeconômicas das populações analisadas e, como não existem estudos semelhantes feitos na população estudada, as comparações tornam‐se menos relevantes. Os resultados são, portanto, de grande importância para o estudo de etiologias em diferentes cenários mundiais.
A predominância de etiologias relacionadas a condições neonatais e causas infecciosas é característica de países em desenvolvimento, onde os investimentos em promoção e prevenção na saúde auditiva são limitados.24 A frequência dessas etiologias em outros estudos varia de 16% a 52%, com a menor prevalência encontrada na Finlândia e a maior na África Subsaariana.3,7,25–27 Neste estudo, a prevalência foi de 28,5%, um valor intermediário quando comparado à média mundial e próximo ao encontrado em outros estudos brasileiros.26,27
As condições relacionadas ao período neonatal (CRPN) foram responsáveis pela perda auditiva em 22,1% dos 140 pacientes avaliados no estudo de Walch et al., 19,8% das perdas auditivas foram decorrentes de condições neonatais,7 enquanto para Morzaria et al. foi de 9,6%.9 Isso poderia ser explicado pelo fato de que nossa amostra inclui muitos pacientes que nasceram em nosso hospital, que é centro de referência para gestação de alto risco em nosso estado.
A prevalência de infecções congênitas foi de 6,4%, possivelmente refletindo melhorias nos métodos de higiene, cuidado pré‐natal e do recém‐nascido, bem como nas campanhas de vacinação. A rubéola, que já foi a causa viral mais comum da perda auditiva,12 teve uma redução significativa após a introdução da vacinação.9 O Brasil teve o último caso registrado em 2009.28
Em contraste com outras infecções congênitas, o CMV tem sido mais estudado nos últimos anos, já que ocupa o primeiro lugar entre as causas virais de deficiência auditiva.12 Em países desenvolvidos, representa a segunda maior causa de perda auditiva em crianças, com uma incidência de 10% a 12%.3,29,30 Em nosso estudo, a maioria dos casos de perda auditiva infecciosa foi devida a CMV e todas as crianças identificadas com infecção congênita por CMV eram sintomáticas, uma vez que a triagem universal para CMV ainda não é recomendada pelas autoridades de saúde no Brasil.
Com o aumento do controle das condições neonatais e doenças infecciosas, além do melhor conhecimento dos genes relacionados à deficiência auditiva, as causas genéticas tornaram‐se a principal etiologia da perda auditiva nos países desenvolvidos, chegam a 50%‐60% dos casos. De acordo com a literatura mundial, a prevalência varia de 15% a 60%.3,7,9,25,31,32 Essa variação pode ser explicada por diferenças no desenvolvimento étnico e socioeconômico da população analisada, bem como pelas variações metodológicas entre os estudos. Em nossa amostra, 22,1% dos casos foram atribuídos à genética e as mutações mais prevalentes foram analisadas, seguiram‐se recomendações relacionadas à investigação de perda auditiva congênita.19,33
A PANS foi responsável pela perda auditiva em 12,9% das crianças estudadas. Costa et al. também avaliaram as causas da perda auditiva em crianças brasileiras e 16% dos casos foram confirmados como genéticos por testes moleculares.26 A etnia tem uma influência significativa na perda auditiva genética não sindrômica. Mutações no gene GJB2, por exemplo, são encontradas predominantemente na Europa, no Norte da África, Oriente Médio e em áreas habitadas por imigrantes desses locais.1,34 A grande maioria dos participantes deste estudo foi de brancos autodeclarados (89,3%), mas é sabido que nossa população apresenta uma importante miscigenação étnica. A miscigenação característica do povo brasileiro começou com índios sul‐americanos, africanos e colonizadores portugueses. Nos séculos 19 e 20, fluxos de imigrantes entraram em território brasileiro e na região Sul a imigração foi principalmente originária da Europa.6
A maioria das mutações deste estudo foi encontrada no gene GJB2, que codifica a conexina 26 (17 das 18 crianças com alterações em testes moleculares). A mutação mais prevalente foi a 35delG, o gene principal foi o GJB2 e dez mutações foram encontradas: três 35delG com expressão bialélica (homozigose) e sete monoalélicas (heterozigose).
A perda auditiva ocorre em pacientes homozigotos e os heterozigotos são chamados de portadores.1 De acordo com a literatura, cerca de 1,35% a 6,5% dos pacientes com perda auditiva são portadores,1,35 com alta probabilidade de a perda auditiva ser de origem genética. Chang et al. descreveram que 2,2% da população com deficiência auditiva são portadores. De acordo com eles, a explicação está na existência de um gene complementar GJB2 ainda não identificado, ou de um fenótipo de portador com penetrância em uma pequena fração dos portadores da mutação GJB2.1 Por essa razão, os cinco casos da mutação 35delG de heterozigotos não variantes identificados neste estudo (3,57% de todas as crianças) foram considerados como casos de perda auditiva não sindrômica.
Duas outras variantes patogênicas do GJB2 também foram encontradas em pacientes heterozigotos para 35delG. Elas foram: c.551G>C (p.Arg184Pro) e c.516G>A (p.Try172*). Neste estudo, a mutação p.Arg184Pro foi encontrada em heterozigose associada à variante 35delG. Essa associação já foi descrita em outros estudos.36,37 O grau de perda auditiva do paciente neste estudo foi profundo, mas Shalev et al. bem como Batissoco et al. descreveram casos com perda auditiva moderada.25,36
A variante c.516G>A (p.Try172*) foi descrita na população brasileira por Oliveira et al., através de dois casos de perda auditiva profunda com a p.Try172* em heterozigose, um deles associado à mutação 35delG.38 Neste estudo, a mesma associação e o mesmo fenótipo foram identificados. Nenhuma outra descrição de p.Try172* foi encontrada, sugeriu que essa variante pode ser típica da população brasileira.
A deleção del(GJB6‐D13S1830), a mais frequente das mutações do gene GJB6 de acordo com a literatura, foi encontrada em uma criança deste estudo. Essas deleções removem todo ou parte do gene GJB6, ou regiões vizinhas a ele, e causam deficiência auditiva quando presentes em homozigose ou em heterozigose em combinação com uma mutação recessiva no gene GJB2. Embora, na maioria dos casos, elas ocorram em combinação com outras mutações no gene GJB2, neste estudo o paciente era homozigoto.39
Como a presença de fatores de risco não excluiria a possibilidade de alteração genética concomitante, optou‐se pela feitura dos testes genéticos em todas as crianças não sindrômicas, independentemente do fato de qualquer outra etiologia ser mais provável. Porém, apenas aqueles sem uma etiologia possível apresentaram alterações nos testes genéticos. Esse resultado levanta a discussão sobre a necessidade de feitura de testes genéticos em todas as crianças não sindrômicas.
A perda auditiva sindrômica foi encontrada em 9,3% do total de crianças do estudo. Considerando apenas o grupo de etiologia genética, 41,93% eram sindrômicas. De acordo com a literatura, a prevalência geralmente fica em torno de 30%.2 A importância desse diagnóstico é que ele conscientiza a assistência à saúde e os pacientes sobre o prognóstico da perda auditiva e, principalmente, as complicações associadas, para que possam ser devidamente tratados. Dos 13 casos identificados, sete ainda não haviam sido diagnosticados, o que é comum. Em muitos casos, é difícil identificar a síndrome à qual pertence o conjunto de diferentes achados. Além disso, há pelo menos 45 genes associados à perda auditiva sindrômica, a maioria dos quais foi descoberto nas últimas duas décadas, mas que não estão disponíveis para teste na maioria dos ambientes clínicos.40
A prevalência de crianças que permaneceram sem diagnóstico etiológico foi de 31,4%. Entretanto, com as mutações dos genes GJB2 e GJB6, 40% das perdas auditivas genéticas não sindrômicas podem ser identificadas.41 Se outros genes causadores de perda auditiva fossem investigados, provavelmente diminuiríamos o número de crianças sem uma etiologia adequadamente definida. Como opção, indica‐se o uso de painéis multigênicos para rastrear um número maior de genes relacionados à perda auditiva. Esses painéis mostraram resultados promissores na redução de custos e aumento do número de genes que podem ser rastreados simultaneamente.42,43
A infecção congênita por CMV é outra possível causa de etiologia para perda auditiva não diagnosticada. A explicação provavelmente está relacionada à alta prevalência dessa infecção e à dificuldade de diagnosticar o recém‐nascido, porque a maioria é assintomática ao nascimento e há uma janela de tempo curta para analisar a carga viral de CMV na urina e/ou saliva.12,29 Por essa razão, a triagem universal para CMV na maternidade tem sido defendida. O diagnóstico precoce dessa infecção congênita é muito importante, pois é uma causa potencialmente tratável de perda auditiva na infância.29
Sobre as etapas do processo investigativo da etiologia da perda auditiva, já era esperado que a maioria dos casos pudesse ser identificada através da história clínica e do exame físico, visto que já se sabia que há uma alta prevalência de perda auditiva relacionada a condições neonatais. Observamos que os testes genéticos foram o segundo maior fator responsável pela definição da etiologia (18,8%), o que confirma a importância da investigação de mutações nas conexinas 26 e 30 em casos de perda auditiva congênita.
Nenhum impacto das diferentes etiologias sobre o grau da perda auditiva foi identificado, o que está em conformidade com os dados da literatura disponível.7,22,31 Entretanto, a alta prevalência de pacientes com perda profunda pode ser um viés em nosso estudo, já que somos um centro de referência em implante coclear.
ConclusãoO predomínio de condições relacionadas ao período neonatal e causas infecciosas são características de países em desenvolvimento e o elevado número de CRPN também pode ser devido ao fato de nosso hospital ser um centro de referência para gestações de alto risco. A baixa prevalência de infecções congênitas possivelmente reflete a melhoria nos métodos de higiene, cuidados pré‐natais e do recém‐nascido, bem como nas campanhas de vacinação, mas acreditamos que o CMV congênito pode desempenhar um papel maior do que é de conhecimento atual. A mutação mais prevalente foi a 35delG, o principal gene foi o GJB2, provavelmente devido à influência europeia no genótipo de nossa população. Cinquenta e um por cento das crianças com deficiência auditiva tiveram a etiologia estabelecida na primeira consulta, através de história clínica e exame físico. Os testes genéticos definiram a causa da deficiência auditiva em 18,8% dos casos.
Conflitos de interesseOs autores declaram não haver conflitos de interesse.
Como citar este artigo: Faistauer M, Lang Silva A, Félix TM, Souza LT, Bohn R, Costa SS, et al. Etiology of early hearing loss in Brazilian children. Braz J Otorhinolaryngol. 2022;88:S33–S41.
A revisão por pares é da responsabilidade da Associação Brasileira de Otorrinolaringologia e Cirurgia Cérvico‐Facial.
gology tem o prazer em homenagear os revisores


